Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Applicazioni genetica umana e molecolare II parte
Dr. Stefania Gonfloni Studio 4319 BIOTECNOLOGIE MOLECOLARI Principi e tecniche Terry A. Brown BIOTECNOLOGIA MOLECOLARE Principi e applicazioni del DNA ricombinante Bernard R. Glick Jack J.Pasternak GENETICA UMANA E MOLECOLARE Stracan T. Read A.P.

2
Che cos’è la clonazione dei geni?
Frammento di DNA con il gene da clonare viene inserito in un Vettore Il vettore trasporta il gene all’interno della cellula ospite Il vettore si moltiplica all’interno della cellula ospite producendo numerose copie uguali non solo a se stesso ma anche del gene che trasporta Quando la cellula ospite si divie, alla progenie vengono passate copie della molecola di DNA ricombinante ed il vettore si moltiplica ulteriormente Dopo un grande numero di divisioni cellulari, si produce una colonia, o clone costituita da cellule identiche.

3
Il procedimento di clonazione del DNA ricombinante
Il procedimento di clonazione del DNA ricombinante. Si separa dall’organismo sorgente un segmento di DNA mediante un’endonucleasi di restrizione e lo si inserisce in un vettore di clonazione. Poi si introduce il costrutto vettore di clonazione-inserto DNA nelle cellule ospiti bersaglio, identificando e coltivando le cellule che portano il costrutto. Se occorre, il gene clonato sarà espresso nella cellula ospite e la sua proteina sarà prodotta e raccolta.

4
Il rilevamento (mappatura dei siti delle endonucleasi di restrizione. A. digestione con endonucleasi di restrizione e separazione elettroforetica dei frammenti. Un segmento di DNA purificato viene reciso separatamente con EcoRI e con BamH1 (digestione individuale) poi con entrambi gli enzimi (digestione duplice). Le righe orizzontali sottostati a ciascuna digestione rappresentano schematicamente la collocazione dei frammenti di DNA (bande) nelle corsie di gel dopo l’elettroforesi e la colorazione del DNA con il bromuro di etidio. I numeri denotano la lunghezza dei prodotti di digestione (frammenti) espresssa in numero di copie di basi. B La mappatura delle endonucleasi di restrizione derivante dalla digestione e dalla separazione elettroforetica illustrate in A.
 poi con entrambi gli enzimi (digestione duplice). Le righe orizzontali sottostati a ciascuna digestione rappresentano schematicamente la collocazione dei frammenti di DNA (bande) nelle corsie di gel dopo l’elettroforesi e la colorazione del DNA con il bromuro di etidio. I numeri denotano la lunghezza dei prodotti di digestione (frammenti) espresssa in numero di copie di basi. B La mappatura delle endonucleasi di restrizione derivante dalla digestione e dalla separazione elettroforetica illustrate in A..")
5
Il vettore di clonazione plasmidico pBR322
Il vettore di clonazione plasmidico pBR322. Siti di riconoscimento esclusivi di HindIII, SalI, BamHi, e PstI sono presenti nei geni per la resistenza alla tetraciclina (Tetr) e all’ampicillina (Amp r). Il sito di riconoscimento esclusivo di EcoRI è collocato appena al di fuori del gene per la resistenza alla tetraciclina. L’origine della replicazione funziona nel batterio di E.coli. La sequenza DNA completa di pBR322 è costituita da 4361 bp.
 e all’ampicillina (Amp r). Il sito di riconoscimento esclusivo di EcoRI è collocato appena al di fuori del gene per la resistenza alla tetraciclina. L’origine della replicazione funziona nel batterio di E.coli. La sequenza DNA completa di pBR322 è costituita da 4361 bp.")
6
La clonazione di DNA estraneo in un vettore plasmidico
La clonazione di DNA estraneo in un vettore plasmidico. Dopo la scissione mediante endonucleasi di restrizione ed il trattamento con la fosfatasi alcalina si lega il DNA plasmidico con il DNA bersaglio digerito con l’endonucleasi di restrizione e si saldano due delle quattro incisure. Questa configurazione molecolare è stabile e le due molecole DNA sono congiunte in via covalente. Dopo l’introduzione nelle cellule ospiti I cicli di replicazione che seguono producono nuove molecole di DNA circolare complete e prive di incisure.

8
Spesso lo scopo degli esperimenti di biologia consiste nell’isolare geni atti a codificare proteine. Creazione e screening di una genoteca. Digestione parziale di un frammento DNA con un endonucleasi di restrizione di tipo II. Di solito le digestione parziali si effettuano variando il tempo o la quantità dell’enzima adoperato. In alcune delle molecole DNA l’endonucleasi di restrizione ha reciso in tutti I siti (marcati ciascuno RE1), mentre in altre molecole si sono verificate meno scissioni. Il risultato desiderato è un campione nel quale le molecole DNA hanno tutte le possibili lunghezze.
, mentre in altre molecole si sono verificate meno scissioni. Il risultato desiderato è un campione nel quale le molecole DNA hanno tutte le possibili lunghezze..")
9
L’ibridazione del DNA. Si denatura il DNA sorgente e si tengono separati I due filamentim solitamente fissandoli ad una matrice solida quale una membrana di nitrocellulosa o di naylon. Si denatura il DNA sonda marcato (spesso lungo da 100 a 1000bp) e lo si aggiunge al DNA sorgente denaturato. In queste condizioni si può verificare l’ibridazione (appaiamento delle basi) tra il DNA sonda e quello sorgente. Si lava la membrana per eliminare il DNA sonda non ibridato e poi la si analizza. Se la sonda si ibrida con il DNA sorgente, la si può rivelare con un saggio che indentifichi il marcatore. Se la sonda non si ibridam non si rivela alcuna marcatore.
 e lo si aggiunge al DNA sorgente denaturato. In queste condizioni si può verificare l’ibridazione (appaiamento delle basi) tra il DNA sonda e quello sorgente. Si lava la membrana per eliminare il DNA sonda non ibridato e poi la si analizza. Se la sonda si ibrida con il DNA sorgente, la si può rivelare con un saggio che indentifichi il marcatore. Se la sonda non si ibridam non si rivela alcuna marcatore..")
10
Lo screening di una genoteca con una sonda DNA marcata (ibridazione di colonie).
.")
11
Lo screening immunologico di una genoteca

12
La PCR Primo ciclo della PCR. Il DNA bersaglio si colloca tra le sequenze 1’ e 2 di un filamento e tra le sequenze 1 e 2’ del filamento complementare. Con il campione si mescolano due inneschi (P1 e P2), e la miscela -che contiene andhe la DNA-polimerasi Taq e I dNTP (deossiribonucleotidi trifosfati) -viene riscaldata a 95C per denaturare il DNA, quindi raffreddata lentamente a 55C. Gli inneschi, presenti in eccesso, si appaiano con il filamento DNA originale del campione durante lo stadio di rinaturazione. Si innalza la temperaturra a circa 75C, e la sintesi del DNA incomincia dall’estremità 3’ delle sequenze innesco e procede oltre le regioni dei filamenti DNA complementari all’altra sequenza innesco. I prodotti di questa reazione sono due lunghi filamenti DNA, che serviranno da stampi nel scondo ciclo di PCR.
, e la miscela -che contiene andhe la DNA-polimerasi Taq e I dNTP (deossiribonucleotidi trifosfati) -viene riscaldata a 95C per denaturare il DNA, quindi raffreddata lentamente a 55C. Gli inneschi, presenti in eccesso, si appaiano con il filamento DNA originale del campione durante lo stadio di rinaturazione. Si innalza la temperaturra a circa 75C, e la sintesi del DNA incomincia dall’estremità 3’ delle sequenze innesco e procede oltre le regioni dei filamenti DNA complementari all’altra sequenza innesco. I prodotti di questa reazione sono due lunghi filamenti DNA, che serviranno da stampi nel scondo ciclo di PCR.")
13
Secondo ciclo della PCR
Secondo ciclo della PCR. La materia prima di questa fase è la miscela di DNA risultante dal primo ciclo PCR. Durante le fasi di denaturazione e di rinaturazione le sequenze innesco si ibridano alle regioni complementari dei filamenti originali e dei filamenti stampo lunghi. La sintesi enzimatica del DNA in vitro produce altri filamenti stamo lunghi dai filamenti originali e altri filamenit stampo brevi dai filamenti stampo lunghi. Lo stampo breve reca una sequenza innesco a una estremità e la sequenza complementare all’altro innesco alla seconda estremità.

14
Terzo ciclo della PCR. Durante la fase di rinaturazione le sequenze innesco si ibridano con le regioni complementari dello stampo lungo originale e con I filamento stampo brevi. La sintesi enzimatica del DNA in vitro produce altri filamenti stampo lunghi dai filamenti originalii e altri filamenti stampo brevi sia dai filamenti stampo lunghi sia dai filamenti stampo brevi.

15
Trentesimo cilco della PCR
Trentesimo cilco della PCR. Giunti al trentesimo ciclo, la popolazione delle molecole di DNA nella provetta è costituita quasi del tutto da filamenti stampo brevi.

16
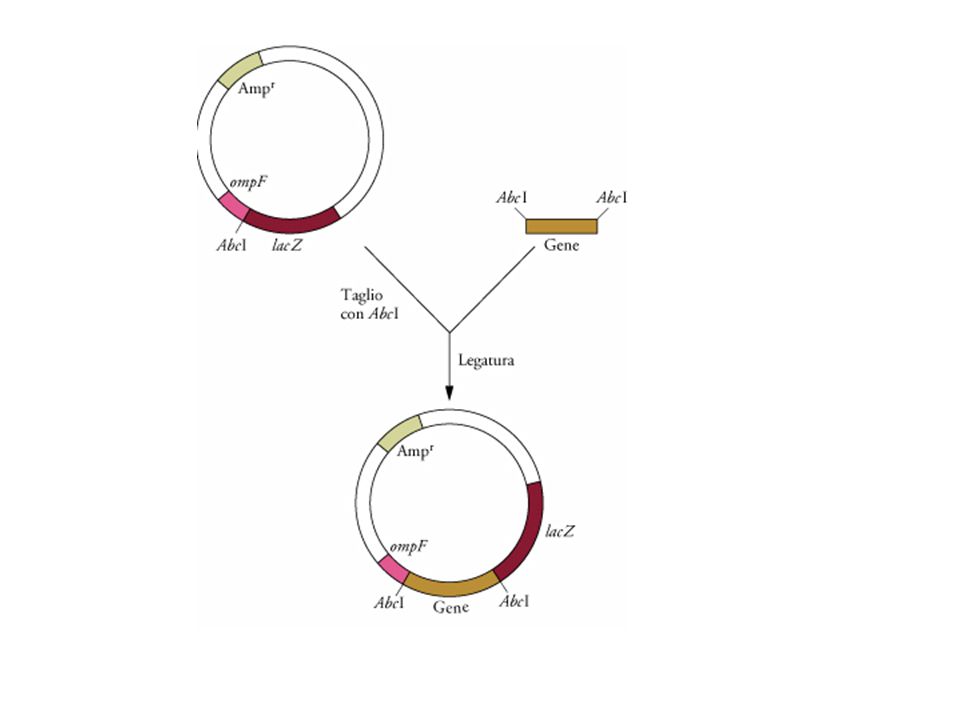
Espressione genica con promotori forti e regolabili
Strategia generalizzata per isolare presunte sequenze promotrici.Si clona in un plasmidio un gene reporter senza promotore. Si recide il DNA cromosomiale con un’endonucleasi di restrizione (AbcI) e lo si clona nel sito Abc1 del plasmidio. Se è presente una sequenza promotrice (p), il gene reporter verrà espresso.
 e lo si clona nel sito Abc1 del plasmidio. Se è presente una sequenza promotrice (p), il gene reporter verrà espresso.")
17
Rappresentazione diagrammatica dell’effetto della concentrazione di glucosio, lattosio, cAMP nel mezzo di coltura sul livello di trascrizione ad opera del promotore lac di E.coli. La freccia indica il verso della trascrizione

18
Doppio sistema plasmidico per controllare il promotore pl di Lambda regolando il repressore cI con il triptofano. Si inserisce in un plasmide, vicino al gene cI, il promotore del triptofano (p trp) e in un secondo plasmide si colloca il promotore pl I posizione adiacente al gene clonato (Gene). Le frecce ondulate denotano la trascrizione. A in assenza di triptofano nel mezzo del gene cI viene trascritto e tradotto e la proteina repressore cI si fissa sul promotore pl bloccando la trascrizione del gene clonato. B in presenza di triptofano il gene cI viene represso, non si crea il prodotto cI e il gene clonato viene trascritto e tradotto.
 e in un secondo plasmide si colloca il promotore pl I posizione adiacente al gene clonato (Gene). Le frecce ondulate denotano la trascrizione. A in assenza di triptofano nel mezzo del gene cI viene trascritto e tradotto e la proteina repressore cI si fissa sul promotore pl bloccando la trascrizione del gene clonato. B in presenza di triptofano il gene cI viene represso, non si crea il prodotto cI e il gene clonato viene trascritto e tradotto..")
19
La scissione delle proteine di fusione ad opera del fattore di coagulazione del sangue Xa. La sequenza di riconoscimento del fattore Xa (sequenza di collegamento di Xa) si colloca tra le sequenze amminoacidiche di due diverse proteine. In seguito alla scissione si libera una proteina funzionale da gene clonato (cona val all’estremo N)
 si colloca tra le sequenze amminoacidiche di due diverse proteine. In seguito alla scissione si libera una proteina funzionale da gene clonato (cona val all’estremo N).")
20
Purificazione cromatografica per immunoaffinita’ di una proteina di fusione. Si ancora ad un supporto solido di polipropilene un anticorpo che si fissa al peptide marcatore della proteina di fusione (anticorpo peptidico anti-marcatore). Si fanno passare le proteine secrete lungo la colonna contenente l’anticorpo legato. La porzione peptide marcatore della proteina di fusione si lega all’anticorpo mentre le altre proteine procedono oltre. La proteina di fusione immunopurificata potrà poi essere selettivamente eluita dalla colonna.
. Si fanno passare le proteine secrete lungo la colonna contenente l’anticorpo legato. La porzione peptide marcatore della proteina di fusione si lega all’anticorpo mentre le altre proteine procedono oltre. La proteina di fusione immunopurificata potrà poi essere selettivamente eluita dalla colonna..")
21
Vettori per l’espressione dei geni
Il vettore di espressione codifica il gene per la resistenza all’ampicillina (Amp r) come gene marcatore selezionabile, per il promotore tac (tac) per il sito di legame del ribosoma lacZ (rbs) per I tre siti di clonazione delle endonucleasi di restrizone (N, P, H) e per due sequenze di arresto della trascrizione (T1 e T2) . La freccia indica il verso della trascrizione. Il plasmide non e’ disegnato in scala. Vettori per l’espressione dei geni ptac: ibrido funzionale dai promotori trp (-35) e lac (-10)
 come gene marcatore selezionabile, per il promotore tac (tac) per il sito di legame del ribosoma lacZ (rbs) per I tre siti di clonazione delle endonucleasi di restrizone (N, P, H) e per due sequenze di arresto della trascrizione (T1 e T2) . La freccia indica il verso della trascrizione. Il plasmide non e’ disegnato in scala. Vettori per l’espressione dei geni. ptac: ibrido funzionale dai promotori trp (-35) e lac (-10)")
22
Integrazione del gene clonato nel sito cromosomico
Due maniere di integrare il gene clonato nel sito cromosomico. A. doppio crossing-over (X-X). B. il gene clonato e’ inserito in posizione adiacente al DNA clonato (c) proveniente dal cromosoma ospite. Tra la regione di DNA del plasmide, c, e quella del cromosoma ospite c’ si verifica l’appaiamento omologo. Un evento unico di ricombinazione (X) nell’ambito della regione di DNA appaiato c-c’ sfocia nell’integrazione dell’intero plasmide, compreso il gene clonato.
. B. il gene clonato e’ inserito in posizione adiacente al DNA clonato (c) proveniente dal cromosoma ospite. Tra la regione di DNA del plasmide, c, e quella del cromosoma ospite c’ si verifica l’appaiamento omologo. Un evento unico di ricombinazione (X) nell’ambito della regione di DNA appaiato c-c’ sfocia nell’integrazione dell’intero plasmide, compreso il gene clonato.")
23
L’inserimento di un gene estraneo in un sito esclusivo predeterminato sul cromosoma di b. subtilis. Nella fase 1 si integra mediante ricombinazione omologa un gene marcatore nel DNA cromosomiale della cellula ospite. Nella fase 2 si sostituisce il gene marcatore con quello bersaglio. Il processo si puo’ poi reiterare con diverse regioni non essenziali del DNA cromosomiale di b. Subtilis b. subtilis

24
Vettore di espressione eucariotico
Vettore eucariotico: una unita’ di trascrizione eucariotica con un promotore (p), un sito di clonazione(cs) per il gene clonato, un segmento di DNA con I segnali di arresto e di poliadenilazione (t) un sistema costituito da un gene marcatore selezionabile eucariotico; un’origine della replicazione che funzioni nella cellula eucariotica (ori euk); un’origine della replicazione che funzioni in E.coli (oriE); un gene marcatore selezionabile in E.coli (Amp r). p:unità di trascrizione eucariotica
, un sito di clonazione(cs) per il gene clonato, un segmento di DNA con I segnali di arresto e di poliadenilazione (t) un sistema costituito da un gene marcatore selezionabile eucariotico; un’origine della replicazione che funzioni nella cellula eucariotica (ori euk); un’origine della replicazione che funzioni in E.coli (oriE); un gene marcatore selezionabile in E.coli (Amp r). p:unità di trascrizione eucariotica.")
25
Le protein ricombinanti prodotte dai sistemi di espressione basati su S. cerevisiae. HIV-1, virus dell’immunodeficienza nell’uomo tipo 1.

26
Metodo per ottenere sequenze di cDNA a lunghezza completa
Metodo per ottenere sequenze di cDNA a lunghezza completa. E’ una forma modificata di RT-PCR che amplifica le sequenze da una regione tra una regione precedentemente caratterizzata nell’mRNA (cDNA) e una sequenza “ancora” accoppiata all’estremità 3’ o all’estremità 5’. Un primeri viene progettato sulla sequenza interna già nota, mentre il secondo primer viene selezionato sulla sequenza “ancorata”. Metodo per ottenere sequenze di cDNA a lunghezza completa
 e una sequenza ancora accoppiata all’estremità 3’ o all’estremità 5’. Un primeri viene progettato sulla sequenza interna già nota, mentre il secondo primer viene selezionato sulla sequenza ancorata . Metodo per ottenere sequenze di cDNA a lunghezza completa.")
27
Saggio di protezione dalla nucleasi S1
Saggio di protezione dalla nucleasi S1. Si sospetta che un frammento di restrizione all’estremità 5’ del gene clonato contenga il sito d’inizio della trascrizione. Esso viene marcato all’estremità 5’, denaturato e mescolato con l’RNA totale ottenuto da cellule in cui si pensa che il gene in questione venga espresso. L’mRNA affine può ibridare con il filamento di DNA antisenso formando un eteroduplex RNA-DNA. Il successivo trattament con la nucleasi S1 dà come risultato il progressivo taglio della sequenza di DNA che sporge in 3’ fino al punto in cui il DNA ibridato con l’estremità 5’ dell’mRNA. B saggio di estenzione del primer in questo caso, viene deliberatamente scelto un piccolo frammento di restrizione che si pensa di contenga il sito di inizio della trascrizione. L’ibridazione con un mRNA affine lascerà l’mRNA con un’estremità che protrude in 5’. Saggio di protezione dalla nucleasi S1

28
SAGE Analisi dell’espressione genica utilizzando il metodo SAGE. Il principio è quello di ridurre ciascuna molecola di cDNA a una corta etichetta di sequenza che lo rappresenti (lunga circa 9 nucleotidi). Le singole etichette vengono poi unite assieme in un unico lungo filamento di DNA, come mostrato in fondo allo schema, dove I numeri sopra la sequenza rappresentano uno specifico cDNA da cui si è tratta l’etichetta.
. Le singole etichette vengono poi unite assieme in un unico lungo filamento di DNA, come mostrato in fondo allo schema, dove I numeri sopra la sequenza rappresentano uno specifico cDNA da cui si è tratta l’etichetta.")
29
Screening multiplo dell’espressione genica
La visualizzazione differenziale dell’mRNA è un metodo rapido per fare uno screening multiplo dell’espressione genica. Visualizzazione differenziale per identificare geni espressi in modo differente In diversi stadi dello sviluppo cardiaco. In questo esempio si mette a confronto l’espressione del cuore di topo a giorni di età embrionale. Sono stati utilizzati tre gruppi di condizioni reazione, con un unico primer arbitrario ed I primer T11A, /t11Co T11G. La figura mostra una porzione del gel dove si possono vedere diverse bande che cambiano d’intensità nei diversi stadi dello sviluppo. E’ da notare un incremento particolarmente marcato nell’espressione di una banda allo stadio di 16 giorni di vita embrionale (indicato dalla freccia).
.")
30
Immunoblotting (Western blotting)
Immunoblottin consiste nell’individuazione di proteine precedentemente separate in base alle dimensioni in un gel di poliacrilammide e nel loro trasferimento (blotting) su una membrana.
 su una membrana.")
31
Espressione della -tubulina in un cervello di un embrione di topo
Immunocitochimica Espressione della -tubulina in un cervello di un embrione di topo di 12, 5 giorni Immunofluorescenza GENE INDICATORE per seguire l’espressione di un gene vivo. L’uso della GFP come etichetta di fusione per seguire la localizzazione delle proteine Proteina a fluorescenza verde come etichetta fornisce un modo utile per seguire l’espressione delle proteine

32
Identificazione di sequenze di regolazione mediante l’uso di geni
Indicatori e le interazioni tra DNA e proteine Le sequenze di regolazione possono essere mappate clonandole in un vettore di espression contenente un gene indicatore privo di promotore. PGL2-basic è un vettore di espressione eucariotico progettato per saggiare le sequenze promotore. Esso contiente un gene indicatore della luciferasi privo di promotore, un segnale di poliadenilazione (poliA di SV40 a valle, un’origine di replicazione convenzionale

33
Analisi delle delezioni nella regionie promotore del gene umano per
Il fattore VIII Analisi delle delezioni nella regione del promotore del gene umano del fattore VIII. Le linee spesse a sinistra indicano sequenze di varie dimensioni a monte del gene del fattore VIII che sono state clonate nel vettore di espressione pGL2-Basic. Le coordinate a -9 fanno riferimento alla metionina di inizio, indicata arbitrariamente come +1.

34
Footprinting mediante DnasiI
Il footprinting mediante Dnasi I identifica le regioni di una molecola di DNA che si legano a proteine, grazie alla loro capacità di conferire resistenza al taglio operato dalla DnasiI. A) Le molecole di DNA sono marcate a una sola estremità. Se il DNA non è complessato con proteine (sinistra), la digestione parziale mediante Dnasi I pancreatica produce una serie di segmenti di DNA di dimensioni differenti perchè la localizzazione di un sito di taglio (trattini blu verticali) su una singola molecola di DNA è essenzialmente casuale.
 Le molecole di DNA sono marcate a una sola estremità. Se il DNA non è complessato con proteine (sinistra), la digestione parziale mediante Dnasi I pancreatica produce una serie di segmenti di DNA di dimensioni differenti perchè la localizzazione di un sito di taglio (trattini blu verticali) su una singola molecola di DNA è essenzialmente casuale.")
35
Saggio di ritardo nel gel
In una molecola di DNA le regioni di legame per le proteine possono essere identificate mediante un saggio di ritardo nel gel e l’indentità della proteina può essere verificata mediante studi di legame competitivo.

36
I sistemi “a doppio ibrido” e a “singolo ibrido” in lievito
A) Il sistema “a doppio ibrido” in lievito. Il gene indicatore integrato nel genoma di lievito verrà espresso solo se è disponibile un adatto fattore di trascrizione. Ciò accade quando vengono messe a contatto due proteine di fusione: una contenente un dominio per il legame al DNA (BD) e l’altra contenente un dominio di attivazione (AD). La proteina bersaglio X è fusa con il dominio di legame che riconosce le sequenze attivatrici a monte (promotore) adiacenti al gene indicatore. La libreria del domino di attivazione rappresentata in altro a destra può esprimere proteine di fusione contenenti un dominio di attivazione associato a una delle molte proteine non caratterizzate (1,2,3,4,). B) il sistema “ a singolo ibrido” in lievito.
 Il sistema a doppio ibrido in lievito. Il gene indicatore integrato nel genoma di lievito verrà espresso solo se è disponibile un adatto fattore di trascrizione. Ciò accade quando vengono messe a contatto due proteine di fusione: una contenente un dominio per il legame al DNA (BD) e l’altra contenente un dominio di attivazione (AD). La proteina bersaglio X è fusa con il dominio di legame che riconosce le sequenze attivatrici a monte (promotore) adiacenti al gene indicatore. La libreria del domino di attivazione rappresentata in altro a destra può esprimere proteine di fusione contenenti un dominio di attivazione associato a una delle molte proteine non caratterizzate (1,2,3,4,). B) il sistema a singolo ibrido in lievito.")
37
Phage display Il phage display è una forma di clonaggio di espressione che comporta il clonaggio di un cDNA in vettori fagici e l’espressione di proteine estranee sulla superficie dei fagi.

38
Preparazione di topi transgenici mediante microiniezione nei pronuclei
Il transgene è presente in tutte le cellule nucleate

39
Le cellule ES modificate geneticamente costituiscono un mezzo per trasferire DNA estraneo o mutazioni specifiche nella linea germinale del topo

40
La mutagenesi mirata mediante ricombinazione omologa puo’
Il metodo del vettore a integrazione. Il DNA vettoriale introdotto (blu) viene tagliato in un unico punto all’interno di una sequenza identica o molto simile a parte di un gene cromosomico (nero). Può succedere che si verifichi ricombinazione omologa (X), portando all’integrazione dell’intera sequenza vettoriale, compreso il gene marcatore (M). Si noti che le lettere non rappresentano degli esoni, ma vogliono semplicemente indicare l’ordine lineare all’interno del gene. B) il metodo del vettore di sostituzione. In questo caso, il gene marcatore è contenuto all’interno della sequenza omologa del gene endogeno e il vettore viene tagliato in un unico punto esterno alla sequenza omologa. Una doppia ricombinazione o un evento di conversione genica (XX) possono dar luogo alla sostituzione di sequenze interne del gene cromosomico con sequenze omologhe del vettore, compreso il gene marcatore. La mutagenesi mirata mediante ricombinazione omologa puo’ inattivare un predeterminato gene cromosomico all’interno di una cellula integra “Hit and run”
 viene tagliato in un unico punto all’interno di una sequenza identica o molto simile a parte di un gene cromosomico (nero). Può succedere che si verifichi ricombinazione omologa (X), portando all’integrazione dell’intera sequenza vettoriale, compreso il gene marcatore (M). Si noti che le lettere non rappresentano degli esoni, ma vogliono semplicemente indicare l’ordine lineare all’interno del gene. B) il metodo del vettore di sostituzione. In questo caso, il gene marcatore è contenuto all’interno della sequenza omologa del gene endogeno e il vettore viene tagliato in un unico punto esterno alla sequenza omologa. Una doppia ricombinazione o un evento di conversione genica (XX) possono dar luogo alla sostituzione di sequenze interne del gene cromosomico con sequenze omologhe del vettore, compreso il gene marcatore. La mutagenesi mirata mediante ricombinazione omologa puo’ inattivare un predeterminato gene cromosomico all’interno. di una cellula integra. Hit and run")
41
La mutagenesi mirata con doppia sostituzione può essere usata
per introdurre nel DNA piccole mutazioni Per introdurre una piccola mutazione senza lasciare sequenze esogene residue, si può utilizzare un metodo a doppia sostituzione con una selezione positiva e negativa. Gli esoni nel gene endogeno sono rappresentati come quadrati numerati, gli introni invece come rettangoli più sottili. Per poter introdurre una piccola mutazione si usa la “conversione genica”. Topi sottoposti a questi riarrangiamenti non possono essere definiti transgenici a causa della mancanza di sequenze estranee nella linea germinale. “Tag and exchange”

42
Il metodo knock-in consente di sostituire l’attività di un gene di un
P promotore, e il sito di poliadenilazione (pA). Il vettore utilizzato per la mutagenesi mirata (knock-in vector) contiene le sequenze clonate del gene En-1, Il metodo knock-in consente di sostituire l’attività di un gene di un dato cromosoma con quella di un altro gene appositamente introdotto
. Il vettore utilizzato per la mutagenesi mirata (knock-in vector) contiene le sequenze clonate del gene En-1, Il metodo knock-in consente di sostituire l’attività di un gene di un. dato cromosoma con quella di un altro gene appositamente introdotto.")
43
Struttura della sequenza di riconoscimento di loxP
Si noti che la sequenza centrale di 8 nucleotidi, fiancheggiata dalle unità ripetute di 13 nucleotidi, è asimmetrica e conferisce un orientamento.

44
Gene trapping Il “gene trapping” utilizza un transgene incapace di essere espresso per selezionare favorevolmente gli eventi di integrazione cromosomica che si verificano dentro o vicino a un gene

45
Clonazione animale La pecora Dolly è il frutto del primo tentativo riuscito di clonazione animale

47
Uso della transgenesi mediata da Yac per produrre un topo con un repertorio di anticorpi umani

Presentazioni simili













 viene copiata in una sequenza complementare di RNA dall’enzima.>")






