Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Disorders of the genome architecture:
clinical diagnosis of constitutional disorders

2
Genetic diseases are recognized to be one of the major categories of human disease
Traditionally genetic diseases are subdivided: chromosomal (numerical or structural aberrations) monogenic or Mendelian diseases multifactorial/polygenic complex diseases epigenetics disorders mitochondrial genetic disorders
 monogenic or Mendelian diseases. multifactorial/polygenic complex diseases. epigenetics disorders. mitochondrial genetic disorders.")
3
With the advent of newer molecular techniques,
a number of new disorders and dysmorphic syndromes are delineated in detail Some of these conditions do not conform to the conventional inheritance patterns and mechanisms are often complex and unique

4
Examples submicroscopic microdeletions or microduplications
trinucleotide repeat disorders epigenetic disorders due to genomic imprinting defective transcription or translation due to abnormal RNA patterning pathogenic association with single nucleotide polymorphisms copy number variations

5
Dhavendra Kumar, Genomic Med., 2009

6
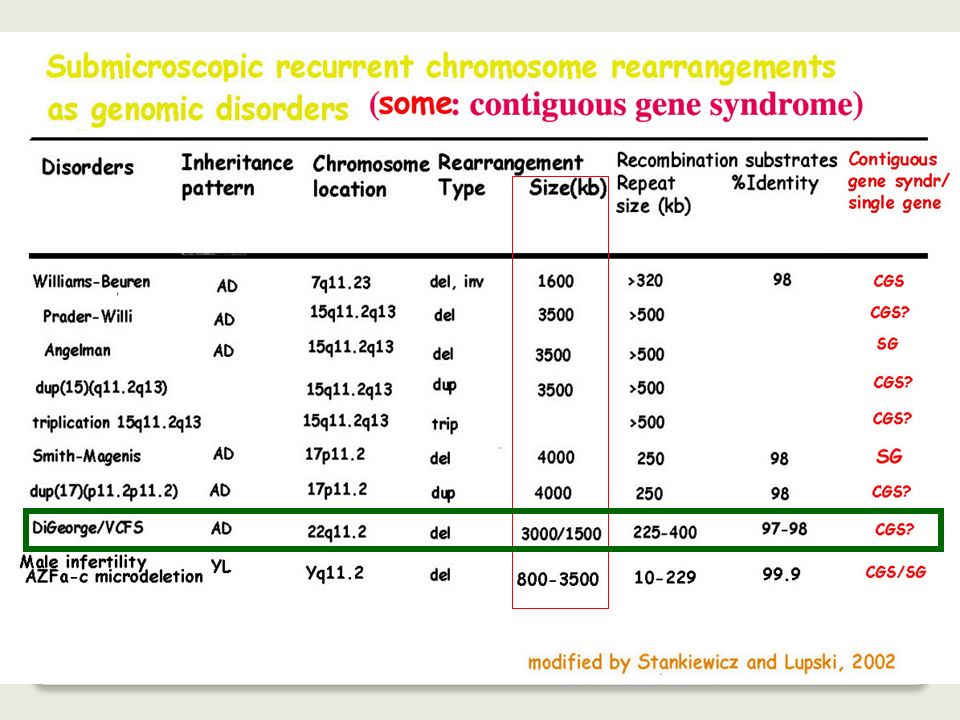
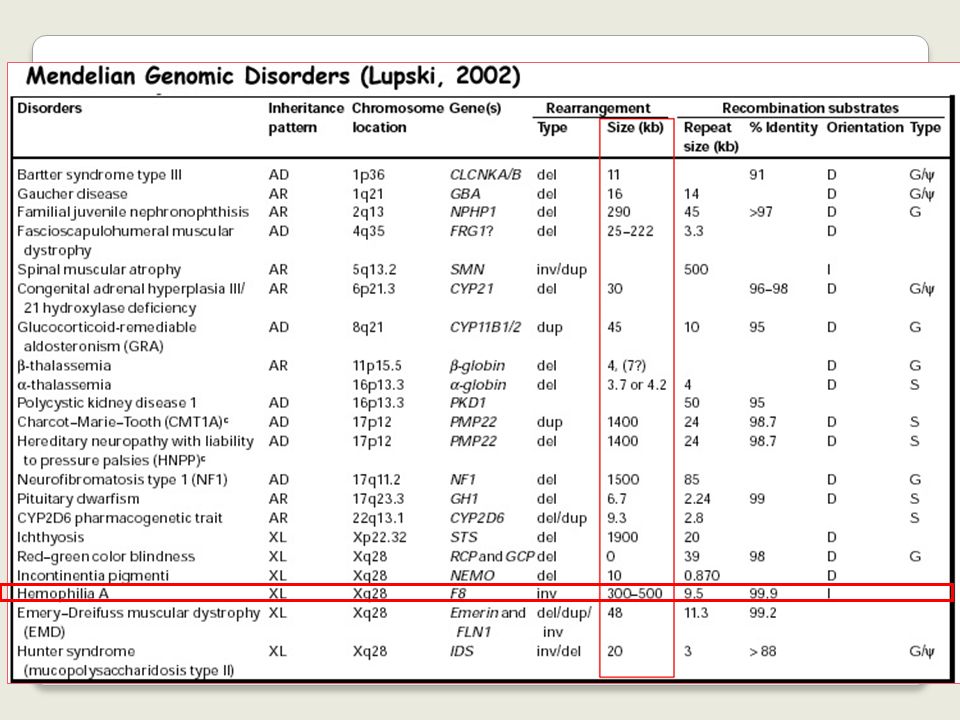
Among these several apparently monogenic disorders result from non-allelic homologous recombination associated with the presence of low copy number repeats on either side of the critical locus or gene cluster.

7
Studi dei breakpoints molecolari sui
riarrangiamenti cromosomici ricorrenti ha reso possibile capire le cause dei riarrangiamenti strutturali

8
When greater than 3 to 5 Mb, such changes are usually
Genomic rearrangements describe gross DNA changes of the size ranging from a couple of hundred base pairs, the size of an average exon, to megabases (Mb) When greater than 3 to 5 Mb, such changes are usually visible microscopically by chromosome studies
 When greater than 3 to 5 Mb, such changes are usually. visible microscopically by chromosome studies.")
9
different from the traditional Watson-Crick base pair alterations
Genomic rearrangements describe mutational changes in the genome such as: duplication deletion insertion inversion translocation different from the traditional Watson-Crick base pair alterations

10
Genomic rearrangements can represent polymorphisms
that are neutral in function, or they can also convey phenotypes via diverse mechanisms: changing the copy number (copy number variation or CNV) of dosage-sensitive genes disrupting genes creating fusion genes other mechanisms
 of dosage-sensitive genes. disrupting genes. creating fusion genes. other mechanisms.")
11
The pathological conditions caused by genomic rearrangements are
collectively defined as GENOMIC DISORDERS

12
Disordini genomici *dovuti a riarrangiamenti ricorrenti del DNA coinvolgenti regioni genomiche instabili **il fenotipo clinico è la conseguenza dei dosaggio anomalo dei geni localizzati nella regione instabile

13
Selected disorders of genome architecture

14
Dhavendra Kumar, Genomic Med., 2009

15
Typically, the term 'genomic rearrangements' is only used
to describe gross DNA changes ranging from thousands to sometimes millions of base pairs that can cover clusters of different genes Monogenic point mutations usually reflect errors of DNA replication and/or repair, whereas the gross genomic rearrangements are often caused by other mechanisms mediated or stimulated by genomic structural features genomic architecture

16
human genome is comprised of region- specific low-copy-repeats (LCRs)
SINEs (short interspersed nuclear elements) The Alu repeat occurs about once every 3 kb in the human genome and includes examples that are transcribed LINEs (long interspersed nuclear elements) HERV (families of human endogenous retroviruses) The completion of human genome project has revealed that about 5% of the human genome is comprised of region- specific low-copy-repeats (LCRs)
 The Alu repeat occurs about once every 3 kb in the human genome and includes examples that are transcribed. LINEs (long interspersed nuclear elements) HERV (families of human endogenous retroviruses) The completion of human genome project has revealed that about 5% of the. human genome is comprised of region- specific low-copy-repeats (LCRs)")
17
Disease-causing genomic rearrangements can be
recurrent, with a common size and fixed breakpoints (breakpoints cluster)
")
18
Recurrent genomic rearrangements Genomic diseases resulting from
recurrent visible genomic rearrangements

19
Disease-causing genomic rearrangements can be non-recurrent with different sizes and distinct breakpoints for each event. The nonrecurrent rearrangements share a common genomic region-of-overlap, the smallest region of overlap (SRO), that encompasses the locus associated with the conveyed genomic disorder
, that. encompasses the locus associated with the. conveyed genomic disorder.")
20
Some of the non-recurrent rearrangements have one of their breakpoints localized in one small genomic region. This grouping of breakpoints is distinct from breakpoint clustering, but like clustering, it may reflect underlying genomic architecture (for example, palindrome or cruciform) important to the rearrangement mechanism
 important to the rearrangement mechanism.")
21
Three major mechanisms have been proposed for genomic rearrangements in the human genome:
Non-allelic homologous recombination (NAHR) Non-homologous end-joining (NHEJ) Fork Stalling and Template Switching (FoSTeS)
 Non-homologous end-joining (NHEJ) Fork Stalling and Template Switching (FoSTeS)")
22
Homologous, non allelic sequences,
which include region-specific repeat gene clusters (“LCRs”), are intimately involved in causing chromosomal rearrangements
, are intimately involved. in causing chromosomal rearrangements.")
23
Lupski: 1991, 1992 Identità di sequenza fra i dupliconi di 24 kb: >95%

24
Among the several genes located in the 1,4 Mb region between
the two CMT1A-REP, only PMP22 is dosage sensitive

25
When the two LCRs are located on the
same chromosome and in direct orientation, NAHR between them causes duplication and/or deletion.

26
When the two LCRs are located on the same chromosome but in opposite orientation, NAHR results in inversion of the fragment flanked by them

27
Interchromosomal and interchromatid NAHR between LCRs in direct orientation result in reciprocal duplication and deletion intrachromatid NAHR creates only deletion

28
Un numero crescente di malattie genetiche è causato
da riarrangiamentiricorrenti del DNA che coinvolgono regioni instabili Questi riarrangiamenti portano a perdita o guadagno di geni sensibili al dosaggio Sono causati dalla presenza di dupliconi che fiancheggiano la regione di delezione o di duplicazione

29
Selected disorders of genome architecture

30
Chromosome 7q deletion and Williams syndrome
(circa 1:20.000) 7 (1,5 Mb) q11.23 N.B.:Non identificabile con citogenetica classica
 7. (1,5 Mb) q N.B.:Non identificabile con citogenetica classica.")
31
: Stenosi aortica sopravalvolare + t(6;7)(p21;q11.23) The elastin gene is disrupted by a translocation associated with suprvalvular aortic stenosis (Curran et al, Cell 73: , 1993)
")
32
The duplicons flanking the Williams-Beuren deletion at 7q11.23
1.5 Mb Identità di sequenza fra i dupliconi : >95%

33
Williams-Beuren: a contiguous gene syndrome
Elastin haploinsufficiency: SVAS (supravalvular aortic stenosis) Mental retardation: ??? Peculiar behaviour: ???
 Mental retardation: Peculiar behaviour:")
35
La delezione 22q11.2 è associata
alla sindrome di DiGeorge/ Velocardiofacial (1:4000 neonati)
")
36
Esiste una facies caratteristica di questa condizione (?):
viso allungato, piramide nasale prominente con radice nasale rilevata e ali ipoplasiche, retrognatia, “hooding” delle palpebre superiori, rime palpebrali sottili e lievemente oblique in alto e in fuori - I disordini del sistema immunitario si manifestano soprattutto nell’infanzia e persistono fino alla giovane età portando a frequenti infezioni respiratorie Con l’età si rendono più evidenti i disturbi dello sviluppo, del comportamento e psichiatrici: ritardo mentale lieve, difficoltà di apprendimento, voce ipernasale, forte impulsività, ansia o fobie

37
Genomic disorders: repeats complexity Chromosome 22q11-specific LCR
(E. Beverly, 2000, 2001)
")
39
La ricombinazione fra i geni e gli pseudogeni “A”
rompe il gene del fattore VIII provocando emofilia di tipo grave A a

40
Il 99% dei casi di inversione del fattore VIII si originano
alla meiosi del nonno materno

41
Genomic diseases resulting from
recurrent visible genomic rearrangements

42
Inv dup(15)
")
43
FISH with PWS/AS probes identifies the type of inv dup(15)
")
44
Normal phenotype Prader-Willi syndrome when associated with 15 UPD or
15q11-q13 deletion Angelman syndrome

45
48,XX,+mar,+mar: normal phenotype

46
Genomic diseases resulting from
recurrent visible genomic rearrangements

47
inv dup(8p): more than 50 cases reported
: more than 50 cases reported")
48
* Hypotonia and development delay * Facial dysmorphism
Inv dup(8p) * Hypotonia and development delay * Facial dysmorphism high forhead broad mouth with inferior prominent lip dysplastic big ears curly hair sparse in temporal area * Corpus callosum agenesis * Severe mental retardation
 * Hypotonia and development delay. * Facial dysmorphism. high forhead. broad mouth with inferior prominent lip. dysplastic big ears. curly hair sparse in temporal area. * Corpus callosum agenesis. * Severe mental retardation.")
49
Floridia et al, 1996: Am J Hum Genet
The breakpoints of inv dup(8p) are remarkably constant Floridia et al, 1996: Am J Hum Genet and inverted
 are remarkably. constant. Floridia et al, 1996: Am J Hum Genet. and inverted.")
50
While building the contigs around the inv dup(8p) breakpoints,
the first not deleted probe gave signals consistent with the olfactory receptors gene clusters localisation

52
Is there anything abnormal in the maternal
apparently normal chromosomes 8?? FISH analysis was performed with probes internal to the REPs

53
In 4 of 12 (33%) families with a proband carrying
the Williams -Beuren deletion, an heterozygous inversion at 7q11.23 was observed in the parent transmitting the disease-related chromosome Osborne et al, Nat Genet 29:321 (2001)
")
54
Osborne, NG, Nov. 2001

55
Alcuni riarrangiamenti cromosomici (quali l’inv dup 8p
e la delezione 7q11.23) considerati de novo (i genitori risultano normali all’analisi citogenetica) si formano a causa di inversioni submicroscopiche presenti nel genitore alla cui meiosi si genera il cromosoma anomalo
 considerati de novo (i genitori risultano. normali all’analisi citogenetica) si formano a causa di inversioni. submicroscopiche presenti nel genitore alla cui meiosi si genera. il cromosoma anomalo.")
56
What about translocations??

57
Y X Alla meiosi I, i cromosomi X e Y ricombinano a livello di PAR1
Regione Pseudoautosomica PAR1 Gene SRY, il primo determinante della differenziazione della gonade in testicolo X Y Alla meiosi I, i cromosomi X e Y ricombinano a livello di PAR1

58
Alla meiosi maschile, la traslocazione fra Xp e Yp
porta alla formazione di 25% di spermatozoi con SRY traslocato all’X e di 25% di spermatozoi con l’Y privo di SRY SRY traslocato su Xp

59
FISH con una sonda del gene SRY in un maschio 46,XX
der(X)t(Xp22.3;Yp11.3) I maschi XX (circa 1:20.000) hanno un cariotipo femminile apparentemente normale. L’analisi mediante FISH con una sonda specifica del gene SRY dimostra la trasposizione della regione distale di Yp su Xp. SRY FISH con una sonda del gene SRY in un maschio 46,XX
t(Xp22.3;Yp11.3) I maschi XX (circa 1:20.000) hanno un cariotipo. femminile apparentemente normale. L’analisi mediante FISH con una sonda specifica. del gene SRY dimostra la trasposizione della regione. distale di Yp su Xp. SRY. FISH con una sonda del gene SRY. in un maschio 46,XX.")
60
submicroscopic inversion, already considered neutral,
Mechanism of the Yp/Xp translocation at the basis of XX males and XY females X Y The polymorphic Yp submicroscopic inversion, already considered neutral, is the preferential background for the PRKX/PRKY translocation

61
La traslocazione fra le porzioni distali di Yp e Xp
avviene quasi sempre alla meiosi di maschi fenotipicamente normali portatori di un’inversione submicroscopica di Yp distale. Tale inversione è un polimorfismo che dà suscettibilità al verificarsi della traslocazione

62
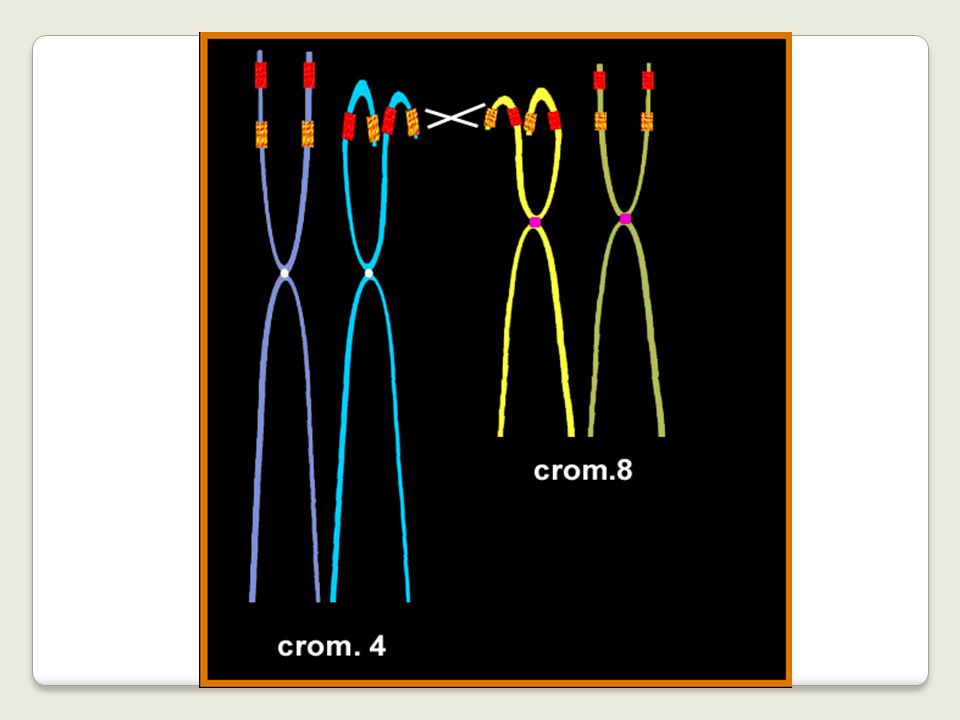
8 4 p16 p23 der(4) der(8)
 der(8)")
63
The t(4;8)(p16;p23) translocation, either in
the balanced or unbalanced form, has been reported in more than 14 de novo or familial cases. Taking into consideration that it may be undetected in routine cytogenetics, it could be the most frequent one after the t(11q;22q) which is itself the commonest translocation in man
 which is itself the commonest translocation in man.")
65
Per definire a livello di sequenza, quali fossero i punti
di rottura dei cromosomi 4 e 8, si è dovuto riempire i “buchi” fra i contigui presenti nelle relative mappe

67
Frequency of the chromosomes 4 heterozygous inversion
in the population: 12% Frequency of the chromosomes 8 heterozygous inversion in the population: 26% Frequency of concurrent chromosomes 4 and 8 heterozygous inversion in the population: 2.5

68
In the last 10 years, seven recurrent submicroscopic inversions have been found:
two pathological, five benign All are mediated by duplicons and those benign represent genomic polymorphisms and give susceptibility to the formation of visible chromosome rearrangements

69
An estimated 5-10% of the human genome consists of
interspersed duplication that have arisen over the past 35 million years of evolution Many regions of the human genome likely contain LCR that predispose to rerrangements

70
Three major mechanisms have been proposed for genomic rearrangements in the human genome:
Non-allelic homologous recombination (NAHR) Non-homologous end-joining (NHEJ) Fork Stalling and Template Switching (FoSTeS)
 Non-homologous end-joining (NHEJ) Fork Stalling and Template Switching (FoSTeS)")
71
non-recurrent chromosome rearrangements?
What about non-recurrent chromosome rearrangements? Non homologous end-joining (NHEJ) can be responsible for many of the non recurrent rearrangements.
 can be responsible for many of the non recurrent rearrangements.")
72
In 21/33 SMS subjects with unusual deletion breakpoints
Evidence for NHEJ has been found by examining breakpoints for some deletions causing SMS In 21/33 SMS subjects with unusual deletion breakpoints the rearrangement occurs in LCR other than the SMS REPs This represents less than 20%–25% of SMS deletion cases Lupski, 2005

73
……..nuove tecniche?????? In presenza di:
ritardo mentale/dismorfismi/anomalie congenite giustifica l’analisi FISH ………………………. …….previo accordo con il genetista/pediatra/neuropsichiatra visto il costo dell’indagine ……consideriamo anche che oggi è possibile eseguire quest’analisi mediante la tecnica MLPA ……..nuove tecniche??????

74
Lo studio delle anomalie cromosomiche “molecular kariotyping”
Gli studi sui telomeri umani e il numero limitato di regioni genomiche note implicate nelle sindromi da microdelezioni dimostrano che microdelezioni e microduplicazioni non visibili con le analisi cromosomiche di routine sono la causa maggiore delle malformazioni umane e del ritardo mentale

75
Lo studio delle anomalie cromosomiche “molecular kariotyping”
Fino a pochissimo tempo fa era impossibile screenare l’intero genoma umano per indivuare la presenza di submicroscopiche alterazioni di tipo numerico Recenti sviluppi nella tecnologia consentono l’analisi genoma per intero con una risoluzione senza precedenti, volte più grande di quella delle tecniche di routine

76
Lo studio delle anomalie cromosomiche “molecular kariotyping”
Lo sviluppo di piattaforme microarray con un alto quantitativo di materiale di lavoro (cialde di vetro o silicone o perline di dimensioni microscopiche sulle quali centinaia di migliaia di sonde di acidi nucleici differenti vengono depositate o sintetizzate in un ordine predefinito) e la disponibilità di differenti format per analizzare specificatamente il DNA, l’RNA o le proteine L’ottimizzazipone dei protocolli comparativi di ibridizzazione e dei sistemi di analisi dei dati Hanno permesso lo sviluppo della cosidetta tecnologia del karyotyping molecolare che consente la rilevazione sensibile e specifica di modificazioni di numero di regioni cromosomiali submicroscopiche lungo l’intero genoma umano
 e la disponibilità di differenti format per analizzare specificatamente il DNA, l’RNA o le proteine. L’ottimizzazipone dei protocolli comparativi di ibridizzazione e dei sistemi di analisi dei dati. Hanno permesso lo sviluppo della cosidetta. tecnologia del karyotyping molecolare. che consente la rilevazione sensibile e specifica. di modificazioni di numero. di regioni cromosomiali submicroscopiche. lungo l’intero genoma umano.")
77
Utilizzi del microarray
Dalla sua introduzione nel 1997, la tecnica del microarray è stata utilizzata per Studi nel campo dell’evoluzione umana (2003) Studio dei tempi di replicazione cellulare (2004) Studio delle variazioni genomiche (2004) Studio dei meccanismi patogentici, di sviluppo e progressione del cancro (2005)
 Studio dei tempi di replicazione cellulare (2004) Studio delle variazioni genomiche (2004) Studio dei meccanismi patogentici, di sviluppo e progressione del cancro (2005)")
78
Utilizzi del microarray
Recentemente (2006……..) lo studio del DNA basato sulla tecnica del microarray è giunto nell’ambito della genetica clinica importante impatto nella diagnosi e nel counseling per i pazienti con ritardo mentale congenito e sindromi malformative, visto che le sue capacità di risoluzione nella ricerca delle anomalie cromosomiche vanno ben al di là di quelle delle tecniche di bandeggio tradizionali. Infatti…
 lo studio del DNA basato sulla tecnica del microarray è giunto nell’ambito della genetica clinica. importante impatto nella diagnosi e nel counseling per i pazienti con ritardo mentale congenito e sindromi malformative, visto che le sue capacità di risoluzione nella ricerca delle anomalie cromosomiche vanno ben al di là di quelle delle tecniche di bandeggio tradizionali. Infatti…")
79
Quando testare i familiari?
Alterazioni numeriche submicroscopiche sia subtelomeriche che interstiziali sono responsabili di RM nel 5-20% dei casi Tali alterazioni submicroscopiche avvengono lungo tutto il genoma umano La frequenza di tali alterazioni congenite è inaspettatamente molto alta Quando testare i familiari? Quante alterazioni sono congenite e quante de novo?

80
Studi pilota: microarray e genetica clinica
…….Ultimi due anni: solo un numero limitato di alterazioni può spiegare la maggior parte delle varietà nella popolazione umana (con un livello di risoluzione di 44 Kbasi) Queste regioni appaiono variare sia nei controlli che nei pazienti Tali sequenze potrebbero essere escluse dai protocolli diagnostici Si riduce considerevolmente il numero delle regioni che spiegano le variazioni di tipo numerico nel genoma umano
 Queste regioni appaiono variare sia nei controlli che nei pazienti. Tali sequenze potrebbero essere escluse dai protocolli diagnostici. Si riduce considerevolmente il numero delle regioni che spiegano le variazioni di tipo numerico nel genoma umano.")
81
Conclusioni: molecular Vs traditional cytogentic technologies
Molecular karyotyping Capacità di individuare alterazioni submicroscopiche causa di malattie DNA isolato direttamente dal sangue o da altro materiale biologico come tessuti bioptici Meccanismi interamente automatizzati, dal laboratorio fino all’analisi dei dati Possibilità di correlare in maniera molto specifica la alterazione genomica con la manifestazione fenotipica Traditional karyotyping Capacità di risoluzione limitata DNA isolato da cellule in replicazione Presenza di procedure manuali, sia in laboratorio che nell’analisi dei dati

82
Conclusioni: molecular Vs traditional cytogentic technologies
Molecular karyotyping Non evidenzia riarrangiamenti bilanciati (inversioni e traslocazioni) Può non rilevare mosaicismi di basso livello Traditional karyotyping Capacità di evidenziare i riarrangiamenti bilanciati Rileva mosaicismi di basso livello Analisi cromosomiche di routine: sempre importanti nella diagnosi di RM e Malformazioni congenite
 Può non rilevare mosaicismi di basso livello. Traditional karyotyping. Capacità di evidenziare i riarrangiamenti bilanciati. Rileva mosaicismi di basso livello. Analisi cromosomiche di routine: sempre importanti nella diagnosi di. RM e Malformazioni congenite.")
83
Array based comparative genome hybridization (array-CGH)
")
84
Usiamo gli enzimi ALUI e RSAI
DIGESTIONE Usiamo gli enzimi ALUI e RSAI MARCATURA Per prima cosa aggiungiamo a ciascun campione 5μl di random primers (kit di marcatura).
.")
85
Old and new genomic disorders

86
Deletion/duplication at 7q11.23
last olig. Normal: 71,865 first olig. Dup: 72,145 last olig. Dup: 73,584 first olig. Normal:73,921 Deletion/duplication at 7q11.23 last olig. Normal: 71,865 first olig. Del: 72,145 last olig. Del: 73,584 first olig. Normal:73,921

87
abnormal cortical development in cortical areas subserving
dup(7)(q11.23): brain MRI a, b: coronal sections; c: axial section abnormal cortical development in cortical areas subserving language function
(q11.23): brain MRI. a, b: coronal sections; c: axial section. abnormal cortical development in cortical areas subserving. language function.")
88
Old and new genomic disorders

89
72.115 72.206 75.837 76.045 MR+ GH deficiency Three cases

90
Homozygous Deletion 17q23.3 of about 40 kb 59.307-59.351 Mb
Patients with IGHD type IA have complete absence of GH The majority of patients with type IA isolated GHD have large deletions within the GH-1 gene Homozygous Deletion 17q23.3 of about 40 kb Mb

91
A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism
Bert B A de Vries, 2006 severe hypotonia from birth long hypotonic face ptosis blepharophimosis large, low-set ears tubular pear-shaped, nose with bulbous nasal tip, long columella with hypoplastic alae nasi broad chin long fingers nasal speech displayed an amiable and friendly disposition

92
Evan E Eichler, Nat Genet, 2006
The individuals had wide ventricles, as assessed by magnetic resonance imaging before the age of 1 year, and in succession appear additional periventricular white matter abnormalities.

93
The 17q21.31 deletion encompasses two known genes, CRHR1 and MAPT and at least two putative genes, IMP5 and STH Gain-of-function mutations in MAPT, encoding the microtubule-associated protein TAU, cause autosomal dominant forms of frontotemporal dementia and parkinsonism

94
Tau-deficient mice showed muscle weakness and memory disturbance
Haploinsufficiency for the microtubule-associated protein TAU may affect axonal elongation and neuronal migration, thereby explaining the major clinical features observed in the 17q21.31 microdeletion positive individuals Tau-deficient mice showed muscle weakness and memory disturbance

95
dup(X)(q28) (400 kb) Normal mother and mildly retarded sister with
last normal: 152, 661 Mb first duplicated: 152,692 Mb last duplicated: 153,130 Mb first normal: 153,148 Normal mother and mildly retarded sister with the same duplication In lymphocytes, skewed X inactivation in the females

96
When to apply array-CGH??
Only to selected cases?? To all mentally retarded subjects requiring cytogenetics analysis??

97
Laura: 22 years old, * Mildly delayed psychomotor development, * Mild broad base gait, * Verbal dyspraxia (poor speech clarity and simple grammatical construction that required a dedicated teacher at school) H 152cm. (10°-25° centile) W 54 (50° centile) CC 56 (90°-97° centile) When 19-years-old WAIS (Wechsler Adult intelligence Scale) test showed verbal IQ= 92; non verbal IQ= 86;
 H 152cm. (10°-25° centile) W 54 (50° centile) CC 56 (90°-97° centile) When 19-years-old. WAIS (Wechsler Adult intelligence Scale) test showed. verbal IQ= 92; non verbal IQ= 86;")
98
Genetic variation in the human genome
Human Molecular Genetics, 2006, Vol. 15, Review

99
Copy-Number Variations
Large segments of the genome, ranging in size from 100 kb to ~3 Mb, varying several folds in copy number in the human population and denominated large-scale copy number variations (LCVs) A higher than expected association between LCVs and known segmental duplications has been noted (Iafrate et al. And Sebat et al. 2005)
 A higher than expected association between LCVs and known segmental duplications has been noted. (Iafrate et al. And Sebat et al. 2005)")
100
CNV influencing gene dosage, expression and disease
Hum Mol Genetics, 2006, Vol. 15, Review

101
It is not clear to what extent such genomic changes are responsible for Mendelian or complex disease traits and common traits (including behavioral traits), or represent only benign polymorphic variation A recent study showed that copy number polymorphism in the FCGR3 gene predisposes to glomerulonephritis in humans and rats Aitman, T.J. Et al, Nature 2006, 439, 851–855

102
Specific categories of genes seem over-represented in CNVs including those important for interaction with the surrounding environment, such as olfaction and response to external stimuli Examples include: glutathione S-transferase genes cytochrome P450 genes the complement component C4 In each case, changes to gene copy number have been shown to give rise to concomitant changes in the level of enzyme activity, with phenotypic consequences. CCL3L1 gene: the increased copy number has been shown to be protective against HIV infection Gonzalez et al, Science 2005

103
Genomic Rearrangements and Phenotypic Traits
Lupski and Stankiewicz, December 2005, Issue 6-e49

104
Genomic Rearrangements and Phenotypic Traits
The two major mechanisms by which variation is introduced into our genome Lupski and Stankiewicz, December 2005, Issue 6-e49 Such variations can be introduced by both endogenous and exogenous means These mutations can cause a disease trait if they affect gene structure, function, or regulation, as well as through the alteration of dosage

105
Il gratta e vinci del DNA
prospettive attuali e future

106
genome-wide analysis had changed genomic medicine from
the hypothesis-driven approaches to the relatively hypothesis-free genetic investigation no more “which one is a good candidate gene for this syndrome?” but “have a look at the entire genome and see what is telling us about these individuals, in this situation”

107
1) Array Comparative Hybridisation (aCGH)
genome-wide analysis: 1) Array Comparative Hybridisation (aCGH) (or SNP Array)
 Array Comparative Hybridisation (aCGH) (or SNP Array)")
108
that cover the entire genome at very high resolution
The array interrogates in a single experiment thousands portions of DNA that cover the entire genome at very high resolution (100 kb, 20 kb, 1 kb, …) balanced rearrangements not detectable
 balanced rearrangements not detectable.")
109
same approach of conventional cytogenetics
but resolution much higher with precise knowledge of gene content at unbalanced regions

110
2) Next Generation Sequencing Massively Parallel Sequencing
genome-wide analysis: 2) Next Generation Sequencing or Massively Parallel Sequencing allows sequencing the entire genome or selected regions (exome sequencing) both balanced and unbalanced rearrangements detectable
 Next Generation Sequencing. or. Massively Parallel Sequencing. allows sequencing the entire genome or selected regions. (exome sequencing) both balanced and unbalanced rearrangements detectable.")
113
Exome sequencing identifies the cause of a mendelian disorder
S. Ng, NatureGenetics November 2009 four affected (Miller syndrome) individuals in three independent kindreds taking into consideration: nonsynonymous variants, splice acceptor and donor site mutations, and coding indels, with both a dominant and a recessive hypothesis filtering against dbSNPS and HapMap polyPhen (exclusion of mutations predicted to be benign)
 individuals. in three independent kindreds. taking into consideration: nonsynonymous variants, splice acceptor and donor site mutations, and coding indels, with both a dominant and a recessive hypothesis. filtering against dbSNPS and HapMap. polyPhen (exclusion of mutations predicted to be benign)")
114
X-chromosome exome sequencing (718 genes: 16 PAR, 702 X-specific) in 208 multiplex XLMR familes
in less than 50% of the families the causative gene had been detected Tarpey et al, NG 2009 3 truncating variants recurrent in controls tr.var.: 10.6% of XLMR

115
what about the remaining 75% of XLMR families???
most but not all X-linked genes have been examined (technical problems) when exome sequencing is negative, other classes of abnormalities have to be searched (regulatory mutations, copy number variants)
 when exome sequencing is negative, other classes of abnormalities have to be searched. (regulatory mutations, copy number variants)")
116
exome sequencing (consumables: 1750 euros)
unrelated affected individuals with largely overlapping phenotypes hypothesizing either AR-disease or AD de novo mutations AD- or X-linked diseases segregating in different members of a family (with complete or incomplete penetrance) AR-diseases when present in more than one member of the family in sporadic cases with genetically heterogeneous diseases for which several genes had already been identified (holoprosencephaly, ambiguous genitalia, epilepsy, retinitis pigmentosa, ……)
 AR-diseases when present in more than one member of the family. in sporadic cases with genetically heterogeneous diseases for which several genes had already been identified (holoprosencephaly, ambiguous genitalia, epilepsy, retinitis pigmentosa, ……)")
117
How exome sequencing might change the diagnostic approach (and medicine)
specialists in different disciplines will be tempted to find the molecular bases of the diseases they are treating categories of patients will be collected not only clinically but also genetically homogeneous so allowing to better define onset and follow-up of that genomic alteration (disease) therapeutic drugs targeted to hit the pathway of altered proteins will be searched asymptomatic relatives will require to know their risk thus promoting research preventing or ameliorating the course of the disease
 therapeutic drugs targeted to hit the pathway of altered proteins will be searched. asymptomatic relatives will require to know their risk thus promoting research preventing or ameliorating the course of the disease.")
118
il gratta e vinci del DNA
have a look at the entire genome and see what is telling us about these individuals, in this situation

Presentazioni simili