Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
TERMODINAMICA STATISTICA I CONCETTI

2
Termodinamica classica
La termodinamica classica fornisce una descrizione delle proprietà macroscopiche della materia (T, P, V, …) e delle loro relazioni a partire da una serie di principi. Principi della termodinamica zeresimo: se A è in equilibrio termico con B e B in eq. termico con C A è in eq. termico con C primo: dU = Q + W secondo: dS = Qrev / T terzo: nello stato a minima energia l’entropia ha un valore ben definito che dipende solo dalla degenerazione dello stato fondamentale.
 e delle loro relazioni a partire da una serie di principi. Principi della termodinamica. zeresimo: se A è in equilibrio termico con B e B in eq. termico con C A è in eq. termico con C. primo: dU = Q + W. secondo: dS = Qrev / T. terzo: nello stato a minima energia l’entropia ha un valore ben definito che dipende solo dalla degenerazione dello stato fondamentale.")
3
Relazioni tra le proprietà macroscopiche.

4
Potenza della termodinamica classica
sulla base di principi di applicabilità universale stabilisce relazioni tra grandezze macroscopiche di sistemi in equilibrio. Es: equazione di Clausius-Clapeyron: ln (p/p0) = H/R (1/T –1/T0) da poche misure di tensione di vapore del solido ( ) e del liquido ( ) a diverse temperature, si ottiene : il calore molare di sublimazione, il calore molare di evaporazione, (da cui il calore molare di fusione), ed il punto triplo di coesistenza delle tre fasi ( ) 1000 K/T ln (P/Torr) -10 -20 -30 Dati sul mercurio
 = H/R (1/T –1/T0) da poche misure di tensione di vapore del solido ( ) e del liquido ( ) a diverse temperature, si ottiene : il calore molare di sublimazione, il calore molare di evaporazione, (da cui il calore molare di fusione), ed il punto triplo di coesistenza delle tre fasi ( ) K/T. ln (P/Torr) Dati sul mercurio.")
5
Potenza … e incompletezza della termodinamica classica
La termodinamica classica è dunque potente …. … ma è incompleta: per ogni elemento, per ogni sostanza, per ogni miscela, esiste un numero irriducibile di parametri termodinamici che non possono essere dedotti dalle equazioni della termodinamica, ma devono essere ottenuti sperimentalmente: Psol (T) Hsub Ttriplo, Ptriplo Hfus Pliq (T) Heva
 Hsub. Ttriplo, Ptriplo Hfus. Pliq (T) Heva.")
6
Incompletezza della termodinamica classica : le Tabelle di dati
Nelle applicazioni termodinamiche sono perciò fondamentali le Tabelle di dati compendio critico di un enorme lavoro sperimentale. Il loro aggiornamento è difficile e poco rimunerativo … … e non sempre è possibile! (Esempi : astrofisica, geologia dell’interno della terra,…) National Institute of Standards and Technology NIST-JANAF THERMOCHEMICAL TABLES, Fourth Edition, Monograph 9 (Part I and Part II) (1998), 1963 pp. by M. W. Chase, Jr.
 National Institute of Standards and Technology. NIST-JANAF THERMOCHEMICAL TABLES, Fourth Edition, Monograph 9 (Part I and Part II) (1998), 1963 pp. by M. W. Chase, Jr.")
7
Utilità (necessità?) di un ponte tra descrizione microscopica (fisica atomica…) e macroscopica (temperatura, entropia…) Perché il punto di fusione dell’oro, Z=79, è 1065 °C… …mentre quello del suo vicino nella tavola periodica, il mercurio, Z=80, è -40 °C ?

8
Termodinamica statistica
La termodinamica statistica fornisce il legame tra le proprietà microscopiche della materia e le sue proprietà macroscopiche. Proprietà di un fluido (Proprietà macroscopiche) Pressione Energia interna Capacità termica Entropia Viscosità Proprietà delle singole molecole Livelli energetici Geometria molecolare Forze intermolecolari Meccanica Quantistica Termodinamica Statistica Classica
 Pressione. Energia interna. Capacità termica. Entropia. Viscosità. Proprietà delle singole molecole. Livelli energetici. Geometria molecolare. Forze intermolecolari. Meccanica. Quantistica. Termodinamica. Statistica. Classica.")
9
Proprietà a livello molecolare
Sperimentali : Spettroscopia Teoriche : Chimica computazionale A partire dalla conoscenza dei livelli energetici delle molecole e delle loro interazioni, la termodinamica statistica permette di ricavare le proprietà macroscopiche.

10
Cenni storici Termodinamica classica Termodinamica statistica
Robert Boyle ( ) Benjamin Thomson, Conte di Rumford ( ) Sadi Carnot ( ) Rudolf Clausius ( ) Termodinamica statistica Josiah Willard Gibbs ( ) Ludwig Boltzmann ( )
 Benjamin Thomson, Conte di Rumford ( ) Sadi Carnot ( ) Rudolf Clausius ( ) Termodinamica statistica. Josiah Willard Gibbs ( ) Ludwig Boltzmann ( )")
11
Tsung-Dao Lee Arrhenius Enrico Fermi Landau Paul Dirac Bose Einstein
James Clark Maxwell Ludwig Boltzmann Josiah Willard Gibbs Yang e Lee : violazione della legge di parità in fisica delle particelle Bardeen: premio Nobel per il transistor e per la teoria della superconduttività Anderson positrone Mott: transizioni metallo-isolante Tsung-Dao Lee Arrhenius Enrico Fermi Landau Paul Dirac Bose Einstein Cheng-Ning Yang Van Hove Langevin Mott Anderson Bardeen …

12
Perché Termodinamica Statistica ?
La meccanica quantistica ci permette di calcolare le proprietà (livelli energetici, …) delle molecole. Le proprietà termodinamiche di massa sono determinate dal comportamento medio di grandi numeri di molecole. Ad esempio la pressione è causata dalla forza media che le collisioni di un gran numero di molecole esercitano contro le pareti del recipiente: non è necessario specificare quali molecole collidono non è necessario considerare le fluttuazioni nel numero di collisioni La termodinamica statistica stabilisce il legame tra proprietà delle singole molecole e proprietà termodinamiche di massa.
 delle molecole. Le proprietà termodinamiche di massa sono determinate dal comportamento medio di grandi numeri di molecole. Ad esempio la pressione è causata dalla forza media che le collisioni di un gran numero di molecole esercitano contro le pareti del recipiente: non è necessario specificare quali molecole collidono. non è necessario considerare le fluttuazioni nel numero di collisioni. La termodinamica statistica stabilisce il legame tra proprietà delle singole molecole e proprietà termodinamiche di massa.")
13
LA DISTRIBUZIONE DEGLI STATI MOLECOLARI
Consideriamo un sistema composto di N molecole. Avvengono innumerevoli collisioni. E’ senza speranza pensare di tener conto delle posizioni, dei momenti, e delle energie interne di tutte le molecole. Il valore di una proprietà macroscopica dipende dal numero di particelle ni in ciascuno stato i. Scambiando l’identità specifica delle particelle in uno stato non cambia il valore della proprietà. PROBLEMA: determinare la popolazione di uno stato, cioè il numero medio di molecole ni che sono nello stato di energia i.

14
L’energia è la somma delle energie delle singole molecole.
Assumiamo che le molecole siano indipendenti, cioè che interagiscono abbastanza per scambiarsi energia durante le collisioni, senza però modificare le energie della molecola isolata. L’energia è la somma delle energie delle singole molecole. Principio di uguali probabilità a priori Tutte le possibilità per la distribuzione di energia sono ugualmente probabili, purché il numero di molecole e l’energia totale siano mantenuti costanti. Cioè assumiamo, per esempio, che gli stati vibrazionali di data energia siano popolati come gli stati rotazionali di ugual energia.

15
Esempio: 4 molecole in un sistema a 3 livelli: le 2 seguenti configurazioni con energia 5 hanno la stessa probabilità. l-l l l l Configurazione: un modo di distribuzione dell’energia. Microstato: una specifica assegnazione dell’energia corrispondente ad una configurazione

16
La probabilità di ogni stato viene valutata semplicemente contando.
Come valutiamo la probabilità di ogni stato ? Per calcolare la probabilità di uno stato particolare, occorre contare il numero di tutti i risultati possibili. Poi occorre contare il numero che dà il risultato desiderato. La probabilità del risultato desiderato è uguale al rapporto tra il numero del risultato desiderato ed il numero totale dei risultati. La probabilità di ogni stato viene valutata semplicemente contando.

17
Coppia di dadi Possibili risultati per una coppia di dadi.
Totale Combinazioni Numero Probabilità % / 3 1+2, / 4 1+3, 3+1, / 5 1+4, 4+1, 2+3, / 6 1+5, 5+1, 2+4, 4+2, / 7 1+6, 6+1, 2+5, 5+2, 3+4, / 8 2+6, 6+2, 3+5, 5+3, / 9 3+6, 6+3, 4+5, / , 6+4, / , / / Somma = 36

19
Sistema costituito da tre particelle distinguibili
Siano tre quanti di energia (ε) disponibili per un totale di energia di 3ε 1 2 3 quanti Energia Come possiamo distribuire l’energia tra le tre particelle?
 disponibili per un totale di energia di 3ε quanti. Energia. Come possiamo distribuire l’energia tra le tre particelle")
20
Prima assegnazione: tutta l’energia ad una particella
1 2 3 1 2 3 1 2 3 Energia 3 modi

21
Seconda assegnazione: 2, 1, 0
3 1 2 3 1 2 3 Energia 1 2 3 1 2 3 1 2 3 6 modi

22
Terza assegnazione 3 Energia 2 1 1 2 3 1 modo

23
Quale è l’assegnazione più probabile ?
3 L’assegnazione con il maggior numero di possibilità In questo caso: “2, 1, 0” 2 3 modi 1 1 2 3 3 Energia 2 6 modi 1 1 2 3 1 2 3 1 modo

24
Supponiamo di avere N molecole in totale.
Configurazioni e pesi Supponiamo di avere N molecole in totale. n0 hanno energia 0, n1 energia 1, n2 energia 2, e così via, dove 0 < 1 < 2 < .... sono le energie dei diversi stati. La distribuzione specifica delle molecole è detta configurazione del sistema, indicata come { n0, n1, n2, } Per esempio, sia un insieme di molecole con 4 stati. Date 19 molecole una configurazione possibile è { 3, 9, 3, 4 }

25
Le configurazioni identificano i modi con cui il sistema può dividere la sua energia complessiva tra gli stati energetici disponibili. Una data configurazione può essere raggiunta in un certo numero di modi. Chiamiamo questo numero di modi W il peso statistico di quella configurazione. Rappresenta la probabilità che questa configurazione possa essere raggiunta in modo puramente casuale a partire da tutte le altre configurazioni. Ciascuna configurazione sarà visitata in maniera esattamente proporzionale al suo peso statistico.

26
la configurazione {3, 2} in quanti modi può essere ottenuta ?
5 molecole la configurazione {3, 2} in quanti modi può essere ottenuta ? Per il secondo livello scelgo la prima molecola tra 5 e la seconda molecola tra le rimanenti 4 Ma scegliere prima la molecola 1 e poi la molecola 2 o prima la molecola 2 e poi la molecola 1 mi porta ad avere lo stesso risultato, la coppia 1,2 nel livello 2 Le restanti 3 molecole vanno nel primo livello in ordine qualsiasi. 5 x 4 / 2 = 10 configurazioni possibili. Una configurazione può avere un numero grande di microstati istantanei.

27
Generalizziamo per un sistema di N particelle che vogliamo distribuire su m livelli.
La prima molecola è scelta tra N, la seconda tra le rimanenti N-1, la terza tra le rimanenti N-2, … fino all’ultima. Ci sono N (N-1) (N-2) = N! modi di scegliere N molecole. Analogamente per le n1 molecole che vanno nel livello 1 ci sono n1! modi di sceglierle, per le n2 molecole che vanno nel livello 2 ci sono n2! modi di sceglierle, … Il numero di modi distinguibili di distribuire un numero totale N molecole n1 nel livello 1 , n2 nel livello 2, … è W = N! / n1! n2! …
 (N-2) = N! modi di scegliere N molecole. Analogamente per le n1 molecole che vanno nel livello 1 ci sono n1! modi di sceglierle, per le n2 molecole che vanno nel livello 2 ci sono n2! modi di sceglierle, … Il numero di modi distinguibili di distribuire un numero totale N molecole n1 nel livello 1 , n2 nel livello 2, … è. W = N! / n1! n2! …")
28
N! differenti modi di disporre N molecole
ni! disposizioni di ni molecole con energia i corrispondono alla stessa configurazione Numero di modi di distribuire 3 oggetti a, b, c in due scatole con disposizione {1, 2}. | a | b c |, | b | c a |, | c | a b |. Ci sono 3 modi = 3! / 1! 2! Si evitano di contare le configurazioni | a | c b |, | b | a c |, | c | b a | attraverso i termini ni! al denominatore

29
{N, 0, 0, } corrisponde ad ogni molecola nello stato fondamentale: c’è un solo modo per ottenere questa configurazione {N-2, 2, 0, } corrisponde a 2 molecole nel primo stato eccitato, ed il resto nello stato fondamentale: può essere ottenuta in N(N-1)/2 modi { n0, n1, n2, } può essere ottenuta in W modi differenti, dove W è detto il peso della configurazione. W può essere calcolato come W = N! / (n0! n1! n2! ...) Calcoliamo la probabilità delle configurazioni semplicemente calcolando in quanti modi possono essere ottenute = numero totale di possibilità diviso per il numero di possibilità equivalenti
/2 modi. { n0, n1, n2, } può essere ottenuta in W modi differenti, dove W è detto il peso della configurazione. W può essere calcolato come. W = N! / (n0! n1! n2! ...) Calcoliamo la probabilità delle configurazioni semplicemente calcolando in quanti modi possono essere ottenute = numero totale di possibilità diviso per il numero di possibilità equivalenti.")
30
{3, 6, 5, 4} Ci sono 18! modi di distribuire 18 molecole in 4 scatole
3! modi di disporre 3 molecole nella prima scatola sono equivalenti 6! modi di disporre 6 molecole nella seconda scatola sono equivalenti ….. Il numero di disposizioni distinguibili è 18! / 3! 6! 5! 4!

31
Numero di modi di distribuire 18 oggetti in 4 scatole secondo la configurazione {3, 6, 5, 4}.
18! / 3! 6! 5! 4! = Anche pochi oggetti generano pesi enormi: calcolarli è un problema. E’ più conveniente lavorare con il logaritmo naturale del peso ln W invece che con il peso W.

32
Approssimazione di Stirling
quando x è grande ln x! x ln x - x x ln x! x ln x – x

33
Applicando l’approssimazione di Stirling

34
Avendo il peso statistico di ogni configurazione, il valor medio di un’osservabile può essere calcolato In pratica una configurazione è dominante. Tale configurazione è la sola da prendere in considerazione quando si calcolano le proprietà medie termodinamiche

35
Siano N molecole distribuite tra 2 stati.
{N, 0}, {N-1, 1}, ..., {N-k, k}, ... , {1, N-1}, {0, N} sono configurazioni possibili ed i loro pesi sono rispettivamente 1, N, ... , N!/(N-k)! k!, ... , N, 1 N=8 {8, 0} W = 1 {7, 1} W = 8 {6, 2} W = 28 {5, 3} W = 56 {4, 4} W = 70 …… {0, 8} W = 1
! k!, ... , N, 1. N=8. {8, 0} W = 1. {7, 1} W = 8. {6, 2} W = 28. {5, 3} W = 56. {4, 4} W = 70. …… {0, 8} W = 1.")
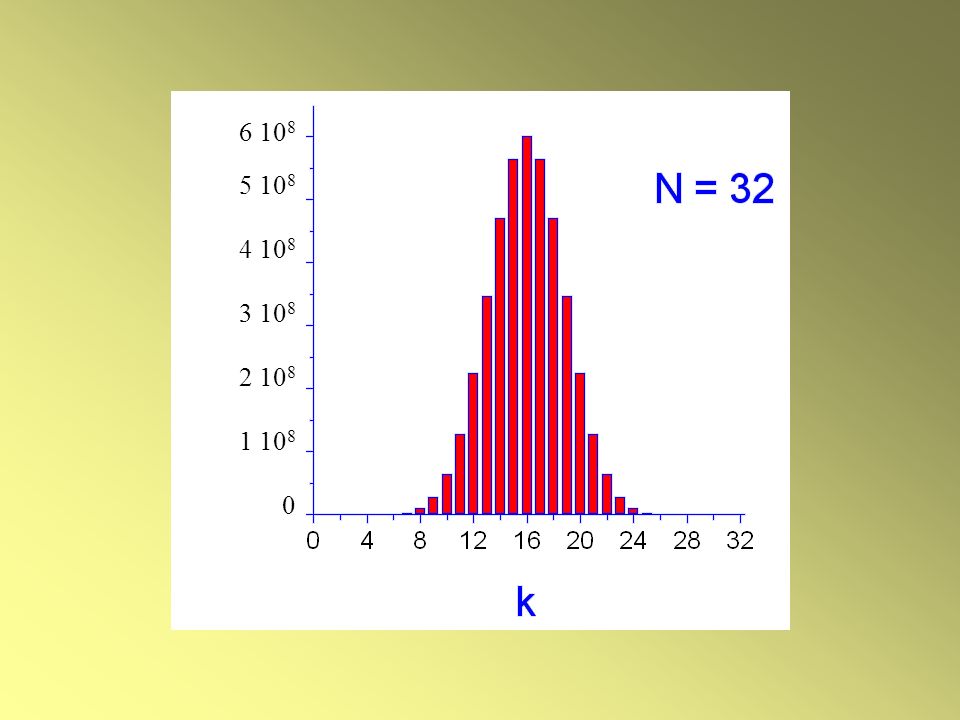
38
6 108 5 108 4 108 3 108 2 108 1 108
39
Quando N è pari, il peso ha il valore massimo a
k = N/2 Wk=N/2 = N! / [N/2)!]2 Quando N è dispari, il peso ha il valore massimo a k = N/2 1 Al crescere di N, il massimo diventa più netto!
!]2. Quando N è dispari, il peso ha il valore massimo a. k = N/2 1. Al crescere di N, il massimo diventa più netto!")
40
R(N) Wk=N/2 / Wk=N/4 = (N/4)! (3N/4)! / [(N/2)!]2
Se mettiamo N/2 molecole nello stato 1 e le rimanenti N/2 nello stato {N/2, N/2} Wk=N/2 = N! / [N/2)!]2 Se mettiamo N/4 molecole nello stato 1 e le rimanenti 3N/4 nello stato 2 {N/4, 3N/4} Wk=N/4 = N! / [(N/4)! (3N/4)!] Il rapporto dei pesi è R(N) Wk=N/2 / Wk=N/4 = (N/4)! (3N/4)! / [(N/2)!]2 | N | | | | | | x 1023 |R(N) | | 2.5 | 7.1 | 57.1 | x 1014 | x 103E+22
![R(N) Wk=N/2 / Wk=N/4 = (N/4)! (3N/4)! / [(N/2)!]2](http://slideplayer.it/slide/588286/2/images/40/R%28N%29+%EF%82%BAWk%3DN%2F2+%2F+Wk%3DN%2F4+%3D+%28N%2F4%29%21+%283N%2F4%29%21+%2F+%5B%28N%2F2%29%21%5D2.jpg "Se mettiamo N/2 molecole nello stato 1 e le rimanenti N/2 nello stato 2 {N/2, N/2} Wk=N/2 = N! / [N/2)!]2. Se mettiamo N/4 molecole nello stato 1 e le rimanenti 3N/4 nello stato 2 {N/4, 3N/4} Wk=N/4 = N! / [(N/4)! (3N/4)!] Il rapporto dei pesi è. R(N) Wk=N/2 / Wk=N/4 = (N/4)! (3N/4)! / [(N/2)!]2. | N | 4 | 8 | 16 | 32 | 256 | 6.0 x |R(N) | 1.5 | 2.5 | 7.1 | 57.1 | 3.5 x 1014 | 2.6 x 103E+22.")
41
Per un sistema macroscopico ( N ~ 1023 ) esiste una configurazione dominante.
I valori istantanei dei numeri di occupazione non sono mai molto diversi dai valori medi. Il sistema in pratica si trova sempre nella configurazione dominante o vicino ad essa (fluttuazioni), cioè in equilibrio: le proprietà del sistema sono quelle caratteristiche della configurazione dominante. Questo risultato non dipende dall’aver considerato 2 stati, vale per qualunque numero di stati. N = stati N = stati {3, 3, 3} W = {5,5,5} W = {5, 3, 1} W = {6,5,4} W = {4, 3, 2} W = {7,5,3} W =
, cioè in equilibrio: le proprietà del sistema sono quelle caratteristiche della configurazione dominante. Questo risultato non dipende dall’aver considerato 2 stati, vale per qualunque numero di stati. N = 9 3 stati N = 15 3 stati. {3, 3, 3} W = 1680 {5,5,5} W = {5, 3, 1} W = 504 {6,5,4} W = {4, 3, 2} W = 1260 {7,5,3} W =")
42
W Wmax Esiste una configurazione dominante {ni}max {ni}

43
…, ma non ci sono molti microstati che danno questi risultati estremi
Ogni microstato è ugualmente probabile Ipotesi fondamentali Se il numero di particelle è grande (>10), il peso della configurazione è dominante
, il peso della configurazione è dominante.")
44
Come determinare la configurazione dominante?
Dobbiamo trovare i valori di ni che massimizzano W o lnW 2 vincoli per il sistema L’energia totale è costante ni i = E = costante Il numero totale di molecole si conserva ni = N = costante Come massimizzare W o lnW con questi vincoli?

45
Quando l’occupazione dello stato i-esimo cambia da ni a ni+dni, la funzione lnW cambia da lnW a lnW + d lnW L’energia totale è costante ini = E idni = 0 Il numero totale di molecole si conserva ni = N dni = 0 Non possiamo porre d lnW = 0 semplicemente ponendo tutte le sue derivate parziali = 0 Per tener conto dei vincoli usiamo il metodo dei moltiplicatori indeterminati di Lagrange

46
Il metodo dei moltiplicatori indeterminati di Lagrange
Si vuole determinare non il minimo assoluto di una funzione F(x,y), ma il minimo soggetto al vincolo C(x,y) = costante F(x,y) vincolo
, ma il minimo soggetto al vincolo C(x,y) = costante. F(x,y) vincolo.")
47
L = F(x1, x2, …, xn) - i i[Ci(x1, x2, …, xn) Costantei]
Un problema di n variabili con k vincoli viene trasformato in un problema di n + k variabili senza vincoli Per minimizzare o massimizzare una funzione F(x1, x2, …, xn) soggetta ai vincoli C1(x1, x2, …, xn) = Costante1 C2(x1, x2, …, xn) = Costante2 . Cm(x1, x2, …, xn) = Costantem i vincoli vengono moltiplicati per una costante ed aggiunti alla funzione F. Si determina il minimo della funzione L L = F(x1, x2, …, xn) - i i[Ci(x1, x2, …, xn) Costantei] L/xi = 0, i=1, 2, ..., n
![L = F(x1, x2, …, xn) - i i[Ci(x1, x2, …, xn) Costantei]](http://slideplayer.it/slide/588286/2/images/47/L+%3D+F%28x1%2C+x2%2C+%E2%80%A6%2C+xn%29+-+%EF%83%A5i+%EF%81%ACi%5BCi%28x1%2C+x2%2C+%E2%80%A6%2C+xn%29+%EF%80%AD+Costantei%5D.jpg "Un problema di n variabili con k vincoli viene trasformato in un problema di n + k variabili senza vincoli. Per minimizzare o massimizzare una funzione F(x1, x2, …, xn) soggetta ai vincoli. C1(x1, x2, …, xn) = Costante1. C2(x1, x2, …, xn) = Costante2. . Cm(x1, x2, …, xn) = Costantem. i vincoli vengono moltiplicati per una costante ed aggiunti alla funzione F. Si determina il minimo della funzione L. L = F(x1, x2, …, xn) - i i[Ci(x1, x2, …, xn) Costantei] L/xi = 0, i=1, 2, ..., n.")
48
Per i vincoli, dCi = 0, quindi dF = 0
DIMOSTRAZIONE dL = dF - i i dCi dL = 0 Per i vincoli, dCi = 0, quindi dF = 0 Esempio: determinare le dimensioni della scatola bidimensionale di area massima e di perimetro 8 F(x,y) = xy y 2x + 2y = C = 2x + 2y – 8 = 0 L = F(x,y) + C = xy + (x + y -4) L/ x = y + = x L/ y = x + = 0 L/ = x + y - 4 = 0 x = y = 2
 = xy y. 2x + 2y = 8 C = 2x + 2y – 8 = 0. L = F(x,y) + C = xy + (x + y -4) L/ x = y + = 0 x. L/ y = x + = 0. L/ = x + y - 4 = 0. x = y = 2.")
49
Costruiamo una nuova funzione L
L = lnW + i ni - i ni i Trovare il massimo di L variando { ni }, , è equivalente a trovare il massimo di W con i due vincoli L/ni = lnW/ni + - i = 0 ln W N ln N - j nj ln nj lnW/ni = (N ln N)/ni - (j nj ln nj)/ ni = - ln (ni/N) -ln (ni / N) + - i = 0
/ni - (j nj ln nj)/ ni = - ln (ni/N) -ln (ni / N) + - i = 0.")
50
Dimostriamo che lnW/ni = - ln (ni/N)
lnW/ni = (N ln N)/ni - (j nj ln nj)/ ni
/ni - (j nj ln nj)/ ni.")
51
Distribuzione di Boltzmann
assicura che la probabilità totale è 1 Distribuzione di Boltzmann L’insieme n0, n1, … per cui W ha il suo massimo valore è dato dalla distribuzione di Boltzmann.

52
La popolazione della configurazione dominante obbedisce alla distribuzione di Boltzmann
La distribuzione di Boltzmann collega le osservabili macroscopiche alle proprietà molecolari microscopiche, ed è capace di spiegare le proprietà di equilibrio di tutti i materiali.

53
Distribuzione di Boltzmann
La più probabile popolazione degli stati del sistema è determinata da un solo parametro. T temperatura termodinamica k costante di Boltzmann k = J K-1 La temperatura termodinamica è il parametro che determina la più probabile popolazione degli stati di un sistema in equilibrio termico.

54
FUNZIONE DI PARTIZIONE MOLECOLARE
Definizione Molecolare: j sono le energie delle singole molecole Se ci sono gi stati con la stessa energia i (il livello energetico i-esimo è g volte degenere), la funzione di partizione q viene riscritta come La somma non è più sugli stati singoli, ma sui livelli energetici Riscriviamo la distribuzione di Boltzmann
, la funzione di partizione q viene riscritta come. La somma non è più sugli stati singoli, ma sui livelli energetici. Riscriviamo la distribuzione di Boltzmann.")
55
Poniamo 0 = 0 Serie infinita che converge tanto più rapidamente quanto più grande è il valore di la spaziatura fra gli stati quantici La convergenza è maggiore a bassa temperatura poiché = 1/kT Quando >> e- 0

56
Se 1 - 0 (Δ) è grande (Δ >> kT)
q (valore minimo di q) Se 1 - 0 (Δ) ≈ kT (energia termica ) q numero grande
 Se 1 - 0 (Δ) ≈ kT (energia termica ) q numero grande.")
57
Interpretazione della funzione di partizione
Se T 0 β q g0 Se T ogni termine 1, q numero totale degli stati (in generale infinito) Funzione di partizione: misura la maniera di “ripartirsi” delle molecole tra i vari livelli
 Funzione di partizione: misura la maniera di ripartirsi delle molecole tra i vari livelli.")
58
E, T E, ∞ E, T=0

59
La funzione di partizione q fornisce un’indicazione del numero medio di stati che sono accessibili ad una molecola alla temperatura T del sistema. A T = 0 solo il livello fondamentale è accessibile. Al crescere di T aumenta il numero di livelli accessibili e per T molto grande virtualmente tutti i livelli sono accessibili. q è un numero puro che può assumere valori che vanno da 1 per T = 0 K (quando solo lo stato fondamentale è accessibile) ad un numero indefinitamente grande al crescere della temperatura. q è una misura di quanto le molecole sono capaci di sfuggire allo stato fondamentale.
 ad un numero indefinitamente grande al crescere della temperatura. q è una misura di quanto le molecole sono capaci di sfuggire allo stato fondamentale.")
60
Funzione di partizione per un oscillatore armonico
ESEMPIO Funzione di partizione per un oscillatore armonico

61
Popolazione dei livelli in funzione della temperatura
T bassa T alta e- e- p p p p

62
Funzione di partizione per un sistema a 2 livelli
ESEMPIO Funzione di partizione per un sistema a 2 livelli 1 2 E=0, g=1 E=ε, g=1 E=0, g=1 E=ε, g=1

63
Popolazioni in un sistema a 2 livelli

64
Demagnetizzazione adiabatica
Entropia S con campo magnetico senza campo Temperatura, T In assenza di campo magnetico l’orientazione degli spin è casuale. Il campo B induce un eccesso di spin : viene ceduta Energia a T costante. Se si toglie il campo in condizioni adiabatiche si ha raffreddamento.

65
Approssimazioni e fattorizzazioni
Funzioni di partizione esatte, analitiche sono rare. Vengono impiegati vari tipi di approssimazioni : livelli di energia densi (quasi continui). stati indipendenti ( fattorizzazione di q). …
. stati indipendenti ( fattorizzazione di q). …")
66
Livelli di energia densi
Funzione di partizione traslazionale Consideriamo il caso mono-dimensionale. Sia una molecola di massa m, libera di muoversi nella direzione x tra x = 0 e x = L. Le energie relative allo stato fondamentale sono

67
Per L grande gli stati formano quasi un continuo
Approssimazioni consideriamo anche n = 0 e sostituiamo n2 – 1 con n2 cresce 1 con le dimensioni della scatola 2 con la massa della particella 3 al crescere di T

68
Fattorizzazione delle funzioni di partizione
Se l’energia è somma di contributi di moti indipendenti, la funzione di partizione q è il prodotto delle funzioni di partizione dei singoli moti. Particella in una scatola di dimensioni X, Y, Z

69
Λ diminuisce al crescere di m e T q cresce con m e V
Λ ha le dimensioni di una lunghezza: lunghezza d’onda termica della molecola. Λ diminuisce al crescere di m e T q cresce con m e V Fissata la massa ed il volume, q cresce con T H K Λ = 71 pm << dimensioni di un recipiente ordinario

70
Meccanica quantistica
Perché la funzione di partizione è così importante ? Meccanica quantistica contiene tutte le informazioni necessarie per calcolare le proprietà di una molecola Termodinamica statistica q contiene tutte le informazioni necessarie per calcolare le proprietà di un sistema di molecole indipendenti

71
RIASSUNTO principio di uguali probabilità a priori
probabilità delle diverse configurazioni valutate semplicemente contando esiste una configurazione dominante, la cui popolazione obbedisce alla distribuzione di Boltzmann la funzione di partizione mette in relazione le osservabili macroscopiche alle proprietà microscopiche molecolari, ed è in grado di spiegare le proprietà all’equilibrio di tutti i materiali.

72
L’ENERGIA INTERNA Relazione tra U e q
L’energia totale (E) di un insieme di particelle distinte ed indipendenti:
 di un insieme di particelle distinte ed indipendenti:")
74
Avendo definito le energie molecolari rispetto allo stato fondamentale 0 = 0, E è l’energia interna relativa al suo valore U(0) a T = 0 Poiché q dipende da altre variabili oltre a T (per esempio V) la derivata rispetto a è in realtà una derivata parziale Per calcolare l’Energia interna U serve solo la funzione di partizione molecolare e la sua dipendenza da T
 la derivata rispetto a è in realtà una derivata parziale. Per calcolare l’Energia interna U serve solo la funzione di partizione molecolare e la sua dipendenza da T.")
75
Energia interna di un sistema a 2 livelli

76
Il valore di Lo ricaviamo per il caso di un gas monoatomico, vale in generale. Il valore dell’energia interna di n moli è Il valore di U per N atomi, calcolato dalla funzione di partizione traslazionale q = V/Λ3, è Confrontando le due espressioni

77
Dimostriamo che ( lnq / )V = - 3/(2)
V = - 3/(2)")
78
L’entropia statistica
Un cambiamento dell’energia interna U può avvenire modificando i livelli energetici i i + di modificando le popolazioni ni ni + dni Lavoro Calore Ridistribuzione della popolazione = q Ridistribuzione dei livelli = w

79
Esempio: particella nella scatola e dipendenza dei livelli da L
Quando del lavoro viene fatto sul sistema, i livelli vengono modificati. Esempio: particella nella scatola e dipendenza dei livelli da L lavoro

80
Quando un sistema è riscaldato a volume costante, i livelli restano invariati, ma le popolazioni cambiano w = 0 calore

81
S = k lnW S = k lnW + costante a T = 0 W = 1
Espressione ricavata in precedenza S = k lnW + costante a T = 0 W = 1 S = k lnW

82
Formula di Boltzmann per l’entropia
S = k lnW dove W è il peso della configurazione più probabile del sistema. La formula di Boltzmann indica che l’entropia è una misura del peso (cioè il numero di modi di ottenere la configurazione di equilibrio), e quindi una misura del disordine collega l’entropia termodinamica macroscopica di un sistema alla sua distribuzione di molecole fra i suoi stati microscopici può essere usata per calcolare l’entropia dalle proprietà microscopiche del sistema è la definizione dell’Entropia Statistica
, e quindi una misura del disordine. collega l’entropia termodinamica macroscopica di un sistema alla sua distribuzione di molecole fra i suoi stati microscopici. può essere usata per calcolare l’entropia dalle proprietà microscopiche del sistema. è la definizione dell’Entropia Statistica.")
84
Entropia statistica ed entropia termodinamica
T 0 W = 1 S terza legge della termodinamica Entropia statistica e funzione di partizione

86
Entropia di un sistema di oscillatori armonici
ESEMPIO Entropia di un sistema di oscillatori armonici

87
Entropia di un sistema a due livelli
ESEMPIO Entropia di un sistema a due livelli 1 2 T 0 W = 1 S 0 T W = 2 S Nk ln 2

88
MOLECOLE INTERAGENTI Ogni sistema ha un insieme di stati quantici che le molecole possono popolare. Questi stati possono adattarsi ad ogni interazione tra le particelle. Trattiamo questi sistemi mediante il concetto di INSIEME

89
INSIEME INSIEME: numero infinito di repliche mentali del sistema di interesse. Sia un sistema chiuso di dato volume (V), composizione (N molecole) e temperatura, replicato volte. Le repliche immaginarie sono in contatto termico (scambio di energia, non di molecole) insieme canonico Canone: secondo una regola
, composizione (N molecole) e temperatura, replicato volte. Le repliche immaginarie sono in contatto termico (scambio di energia, non di molecole) insieme canonico. Canone: secondo una regola.")
90
Insiemi Statistici Microcanonico Canonico Macrocanonico
nessuno scambio scambio di energia scambio di energia e massa E costante T costante μ costante

91
CONFIGURAZIONE DOMINANTE
Le repliche hanno energia totale Ciascuna ha energia Ei. Ci sono repliche con energia Ei. Configurazione e peso W dell’insieme. Esiste una configurazione dominante. Proprietà termodinamiche come media sull’insieme usando la singola configurazione dominante. Per analogia Fissati ed , la configurazione di peso più grande è data dalla funzione di distribuzione canonica Q funzione di partizione canonica

92
Numero di stati Fluttuazioni rispetto alla distribuzione più probabile
La distribuzione canonica, che dà la probabilità di uno stato con energia Ei, è solo apparentemente una funzione esponenzialmente decrescente dell’energia Numero di stati Ampiezza dell’intervallo Energia Densità degli stati = numero di stati in un dato intervallo di energia / ampiezza dell’intervallo. La densità degli stati è una funzione rapidamente crescente dell’energia.

93
Il numero di stati cresce esponenzialmente con E.
La distribuzione dei membri di un insieme canonico è data dal prodotto della probabilità di trovarsi nello stato di energia Ei, una funzione esponenzialmente decrescente dell’energia, , per il numero di stati con energia Ei, una funzione rapidamente crescente. Probabilità dello stato dell’energia Numero di stati Energia Il numero di stati cresce esponenzialmente con E. La probabilità che uno stato sia popolato decresce esponenzialmente con Ei. Quindi la distribuzione complessiva dell’energia è una funzione con un picco molto stretto. Quasi tutti i membri dell’insieme hanno un’energia uguale all’energia media.

94
L’informazione termodinamica della funzione di partizione canonica Q
L’energia interna Se l’energia totale dell’insieme contenente membri è La frazione di membri dell’insieme nello stato con energia Ei è

95
L’entropia Il peso totale di una configurazione è il prodotto del peso medio di ciascun membro.

96
Molecole indipendenti
Se le molecole sono indipendenti, l’Energia è la somma delle energie delle singole molecole. In ciascuno degli stati i dell’insieme, ogni particella (1, 2, 3, …) sarà in uno dei possibili stati molecolari j (a, b, c, d, …) una sola volta in ciascuno stato del sistema. La funzione di partizione canonica è:
 sarà in uno dei possibili stati molecolari j (a, b, c, d, …) una sola volta in ciascuno stato del sistema. La funzione di partizione canonica è:")
97
Se fattorizziamo ciascuna particella dalla sommatoria sugli stati del sistema e raccogliamo tutti i termini che si riferiscono ad una data particella, si ottiene: Particelle distinguibili Se tutte le molecole sono dello stesso tipo e distinguibili (per esempio per la loro posizione in un reticolo cristallino), non occorre l’indice per la molecola
, non occorre l’indice per la molecola.")
98
Particelle indistinguibili
Se le particelle sono indistinguibili (per esempio molecole in un gas) il numero di stati accessibili è minore rispetto al caso di particelle distinguibili. Il membro i dell’insieme differisce dal membro j dell’insieme se le particelle sono distinguibili, a causa dell’interscambio delle particelle 2 e 3 tra gli stati b e h. Tuttavia, i è identico a j se le particelle sono indistinguibili.
 il numero di stati accessibili è minore rispetto al caso di particelle distinguibili. Il membro i dell’insieme. differisce dal membro j dell’insieme. se le particelle sono distinguibili, a causa dell’interscambio delle particelle 2 e 3 tra gli stati b e h. Tuttavia, i è identico a j se le particelle sono indistinguibili.")
99
E E Per molecole indipendenti distinguibili :
Per molecole indipendenti indistinguibili :

Presentazioni simili


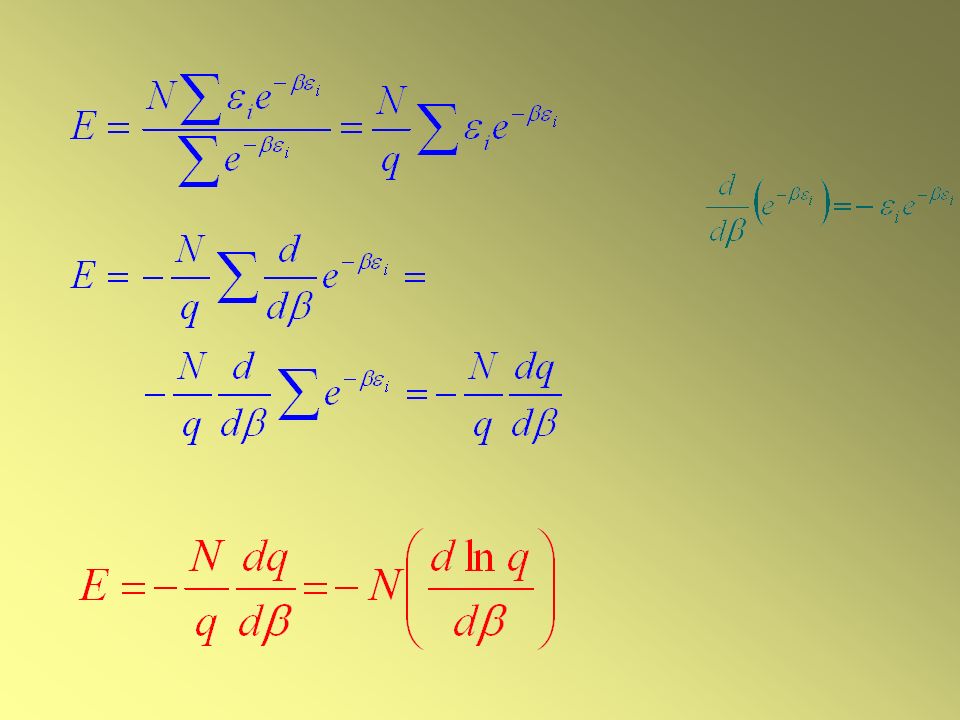

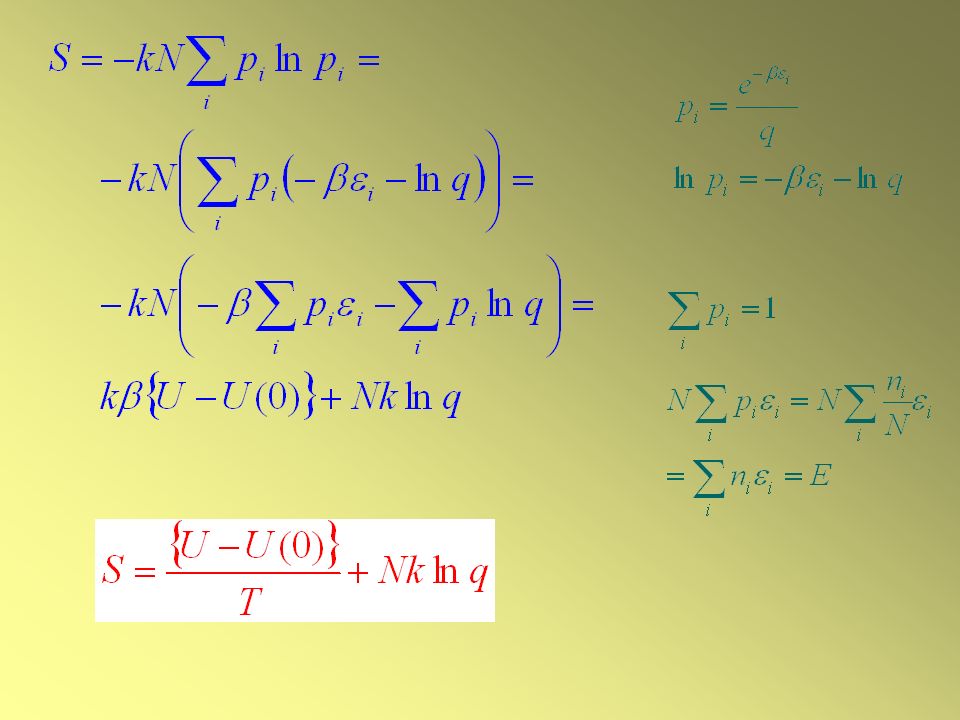











 Acidi poliprotici>")







>")