Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Elettroforesi nel laboratorio di proteomica
Università degli Studi di Perugia Corso di Laurea Interfacoltà in Biotecnologie Fondamenti di Bioinformatica e di Biologia dei Sistemi Proteomica e Metabolomica Elettroforesi nel laboratorio di proteomica Pier Luigi Orvietani Biotecnologie

5
LE PROTEINE HANNO FUNZIONI BIOLOGICHE DIVERSE:
ENZIMI PROTEINE DI TRASPORTO PROTEINE DI RISERVA PROTEINE CONTRATTILI O MOTILI PROTEINE STRUTTURALI PROTEINE DI DIFESA PROTEINE REGOLATRICI ECC.

6
DALLA SEQUENZA ALLA STRUTTURA:
La funzione di una proteina dipende dalla sua sequenza aminoacidica

9
I FOGLIETTI BETA SONO STRUTTURE ESTESE CHE TALVOLTA FORMANO “BARILI”

10
La condensazione di elementi multipli di struttura secondaria porta alla
STRUTTURA TERZIARIA Strutture della trioso fosfato isomerasi e dididro folato reduttasi: elementi di struttura secondaria simili ma diversa struttura terziaria

11
PROTEINE INTEGRALI DI MEMBRANA
Si tratta di proteine inserite in un ambiente idrofobico Le alfa-eliche sono l’elemento di struttura secondaria più comune.

12
DALLA STRUTTURA ALLA FUNZIONE
Esistono molti livelli di funzione delle proteine, che si articolano dalle semplici riorganizzazioni atomiche fino ai cambiamenti nello sviluppo di un organismo Tutti coinvolgono il legame con altre molecole Il riconoscimento molecolare è alla base del funzionamento di una proteina Un esempio è l’attività catalitica Biologia strutturale: capire la funzione attraverso la struttura

13
LE FUNZIONI DI TUTTE LE PROTEINE DIPENDONO DALLA LORO CAPACITA’ DI LEGARE ALTRE MOLECOLE : LIGANDI
INTERAZIONI LIGANDI-PROTEINE: INTERAZIONI NON COVALENTI TRA SUPERFICIE PROTEICA E LIGANDO VARIAZIONI DI CONFORMAZIONE DELLA PROTEINA: POSSONO CONSENTIRE IL LEGAME POSSONO FAVORIRLO POSSONO IMPEDIRLO IL RICONOSCIMENTO MOLECOLARE DIPENDE DA MICROAMBIENTI SPECIALIZZATI CHE DERIVANO DALLA STRUTTURA TERZIARIA DELLA PROTEINA

15
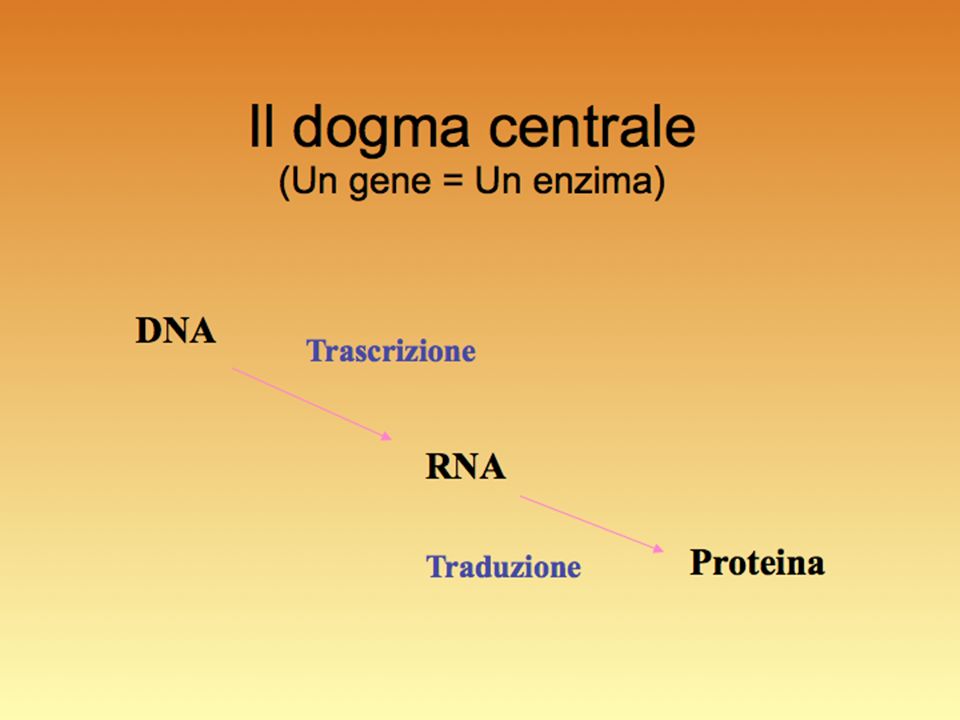
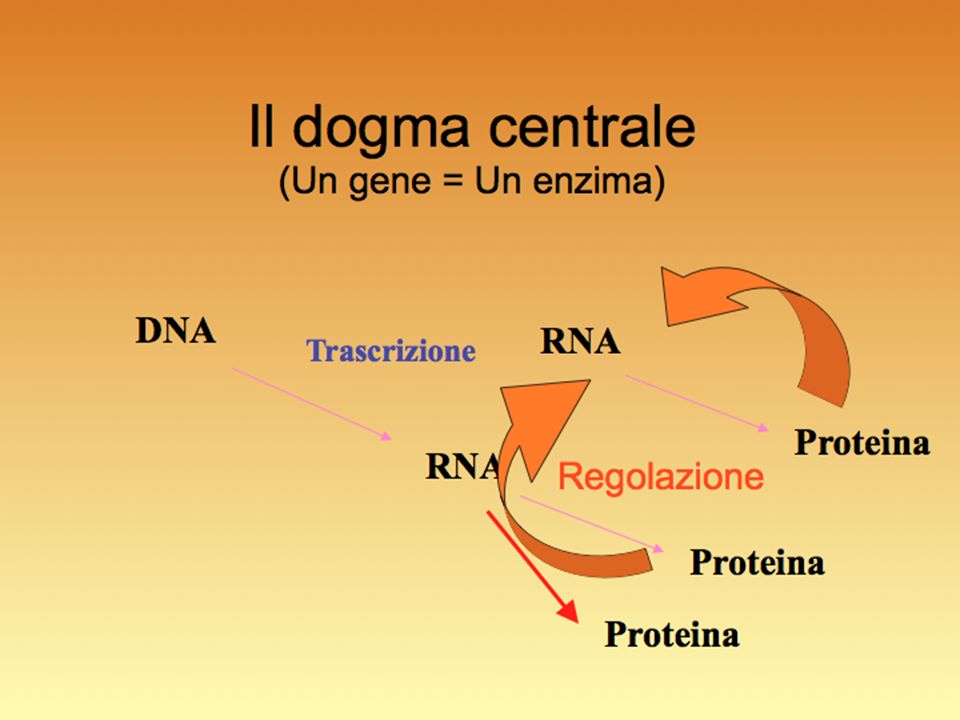
Espressione genica: il risultato finale della trascrizione + processamento dell’RNA + sintesi della proteina e suo funzionamento, cioè il fenotipo.

18
GRANDE FAMIGLIA DEGLI “OMA”
PROTEOMICA: studia tutte le proteine espresse in un certo momento, incluse le isoforme e le modifiche post-traduzionali TRASCRITTOMICA: studia l'insieme degli mRNA trascritti nell'intero organismo, tessuto, cellula METABOLOMICA: studia la totalità dei metaboliti presenti in un organismo LIPIDOMICA: studia la totalità dei lipidi INTERATTOMICA: studia la totalità delle interazioni molecolari che hanno luogo in un organismo; un nome che comunemente indica la disciplina della interattomica è quello di biologia dei sistemi (system biology). SPLICEOMICA: studia la totalità delle isoforme proteiche dovute a splicing alternativo Ecc.
. SPLICEOMICA: studia la totalità delle isoforme proteiche dovute a splicing alternativo. Ecc.")
19
LA PROTEOMICA La proteomica è l’insieme delle tecnologie e degli approcci sviluppati per lo studio del proteoma PROTEOMA: il termine proteoma, ormai universalmente accettato come equivalente linguistico di genoma, fu coniato da M.R. Wilkins nel 1994 per definire il complemento proteico codificato da un genoma Oggi la definizione di proteoma può essere ampliata considerando i seguenti punti: identifica tutte le proteine prodotte in una determinata cellula, tessuto o organismo; definisce come queste proteine interagiscono tra loro descrive la precisa localizzazione delle proteine all’interno della cellula descrive infine l’esatta struttura tridimensionale di queste proteine (lo scopo è quello di trovare il loro punto debole, ossia il punto in cui l’azione di un farmaco potrebbe attivarne o disattivarne la funzione)
")
20
PERCHE’ STUDIARE IL PROTEOMA?
Il sequenziamento del DNA umano (progetto genoma) e lo studio dell’espressione dell’mRNA (microarray) non forniscono tutte le informazione necessarie per lo studio dei processi biologici: malattie (2% delle malattie dell’essere umano sono caratterizzate dal difetto di un singolo gene) differenziamento invecchiamento effetti dell’ambiente La diversità, la funzionalità e l’abbondanza proteica dipendono infatti anche da altri meccanismi (regolazione della traduzione, folding, modificazioni post-traduzionali e degradazione proteica) che regolano l’espressione genica. La proteomica permette di studiare l’espressione proteica di una cellula o tessuto in un determinato momento, perché prende in considerazione il prodotto finale dell’espressione genica, cioè le proteine.
 e lo studio dell’espressione dell’mRNA (microarray) non forniscono tutte le informazione necessarie per lo studio dei processi biologici: malattie (2% delle malattie dell’essere umano sono caratterizzate dal difetto di un singolo gene) differenziamento. invecchiamento. effetti dell’ambiente. La diversità, la funzionalità e l’abbondanza proteica dipendono infatti anche da altri meccanismi (regolazione della traduzione, folding, modificazioni post-traduzionali e degradazione proteica) che regolano l’espressione genica. La proteomica permette di studiare l’espressione proteica di una cellula o tessuto in un determinato momento, perché prende in considerazione il prodotto finale dell’espressione genica, cioè le proteine.")
21
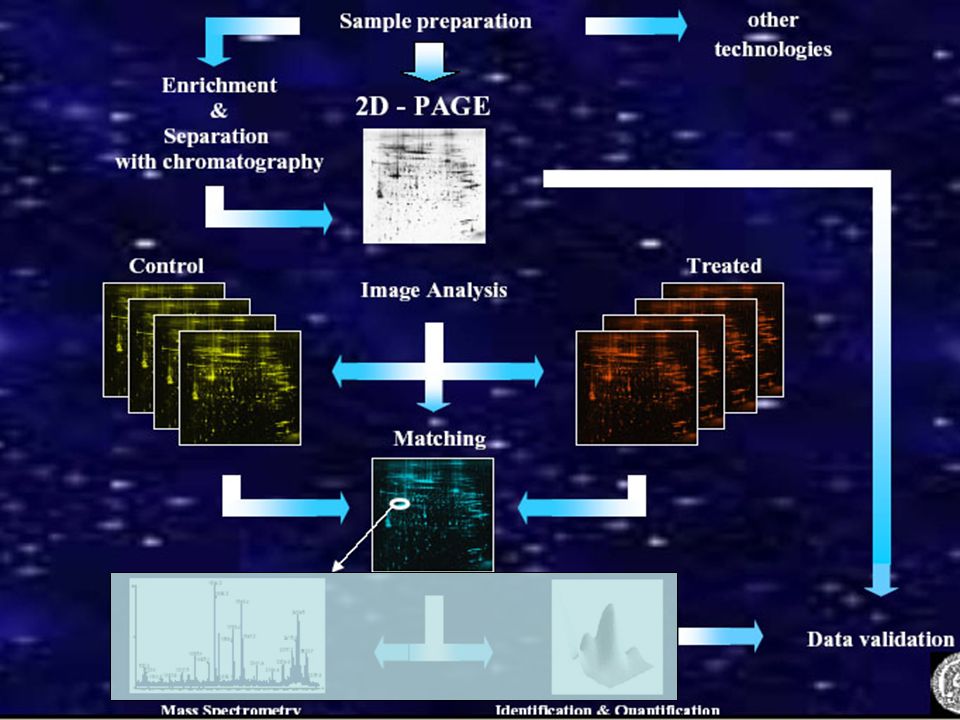
Separare le proteine all’interno del Proteoma
La Proteomica ha come scopo base lo studio del proteoma. Obiettivi della Proteomica sono l’identificazione e l’eventuale sequenziamento delle proteine. Identificare vuol dire saper riconoscere una proteina , mentre il sequenziamento è una procedura che ha lo scopo di determinare l’esatta sequenza peptidica di una proteina Separare le proteine all’interno del Proteoma Un metodo di separazione proteica è l’Elettroforesi

22
2D-Elettroforesi

23
2-D PAGE o BIDIMENSIONALE
Isoelectric focusing (IEF) SDS- Page PUNTO ISOELETTRICO GRANDEZZA MOLECOLARE Si basa essenzialmente sull’applicazione di due tecniche già da tempo conosciute
 SDS- Page. PUNTO ISOELETTRICO. GRANDEZZA MOLECOLARE. Si basa essenzialmente sull’applicazione di due tecniche già da tempo conosciute.")
24
Potenzialita’ della 2-DE
Grande capacità di separazione e risoluzione di miscele proteiche( proteine)ad elevato ordine di grandezza • Risoluzione estremamente alta • I gel 2 –DE sono collettori di frazioni proteiche molto efficienti • Proteine sono protette all’interno della matrice del gel Possibilità di confronto tra i dati Riproducibilità e affidabilità dei dati Capacità di rilevazione di modificazioni post-traduzionali delle proteine glicosilazione fosforilazione tagli proteolitici Possibilità di costruire o di usufruire di mappe di riferimento disponibili in rete
ad elevato ordine di grandezza. • Risoluzione estremamente alta. • I gel 2 –DE sono collettori di frazioni proteiche molto efficienti. • Proteine sono protette all’interno della matrice del gel. Possibilità di confronto tra i dati. Riproducibilità e affidabilità dei dati. Capacità di rilevazione di modificazioni post-traduzionali delle proteine. glicosilazione. fosforilazione. tagli proteolitici. Possibilità di costruire o di usufruire di mappe di riferimento disponibili in rete.")
26
campioni da analizzare
- Fluidi biologici (sangue, urine, saliva, CFS ecc.) - Tessuti Trattati sempre a 4°C - Cellule Congelati in azoto liquido e Stoccati in aliquote in azoto liquido o tenute a – 80°C Una delle cose fondamentali per un buona separazione in 2DE è un giusto prelievo con poche manipolazioni Congelare immediatamente vuol dire fotografare in quel momento l’assetto proteico del campione da analizzare
 - Tessuti. Trattati sempre a 4°C. - Cellule. Congelati in azoto liquido e Stoccati in aliquote in azoto liquido o tenute a – 80°C. Una delle cose fondamentali per un buona separazione in 2DE è un giusto prelievo con poche manipolazioni. Congelare immediatamente vuol dire fotografare in quel momento l’assetto proteico del campione da analizzare.")
27
del quale agisce un pestello di teflon o di vetro (Potter).
Metodi di distruzione delle cellule e dei tessuti frullatori : sono utilizzati per sospensioni di cellule vegetali o animali, o per pezzi di tessuto. omogenizzatori : il tessuto viene messo in un provettone di vetro all'interno del quale agisce un pestello di teflon o di vetro (Potter). Le membrane sono lacerate dalle forze frizionali che il pestello esercita contro le pareti del cilindro di vetro. che può essere azionato a mano o mediante un motore elettrico
. Le membrane sono lacerate dalle forze frizionali che il pestello esercita contro le pareti del cilindro di vetro. che può essere azionato a mano o mediante un motore elettrico.")
28
Sonicazione: ideale per sospensioni cellulari, onde sonore ad alta frequenza (> 20KHz) x 30”- 60” > > distruzione cellule per forze taglianti e cavitazionali (compressione e rarefazione dovuta a formazione e scoppio bolle). Per volumi “piccoli” ( max 100 ml) aggiungendo ghiaccio (sviluppa calore). La strumentazione utilizzata può essere di due tipi: Bagnetto sonicatore Sonda sonicatrice
 aggiungendo ghiaccio (sviluppa calore). La strumentazione utilizzata può essere di due tipi: Bagnetto sonicatore. Sonda sonicatrice.")
29
Metodi per diminuire tali interferenze
Per una buona separazione in 2DE il campione deve essere il più possibile puro e privo di contaminanti DNA Lipidi Sali detergenti polisaccaridi Disturbano il processo di isoelettrofocalizzazione e quello di colorazione del gel. Metodi per diminuire tali interferenze A volte, durante la preparzione del campione, a seconda del tipo di mezzo estrattivo e della particolare procedura utilizzata si ha il campione contaminato da sostanze che interferiscono con l’analisi in prima dimensione (IEF). Concentrazione con taglio molecolare(cut-off) Prepurificazione cromatografica Precipitazione
. Concentrazione con taglio molecolare(cut-off) Prepurificazione cromatografica. Precipitazione.")
30
PRECIPITAZIONE La solubilità delle proteine è il risultato di interazioni polari dei loro residui amminoacidici di superficie con le molecole di solvente acquoso, interazioni ioniche con i sali presenti e varie forze repulsive tra molecole dotate di identica carica Sono 5 gli agenti comunemente usati per far precipitare le proteine: sali inorganici pH e temperatura solventi organici proteine basiche polietilenglicole Se tali interazioni vengono meno, le molecole proteiche interagiscono prevalentemente tra di loro formando aggregati che precipitano. La facilità o difficoltà con la quale la proteina viene impossibilitata a interagire col solvente dipende largamente dalla natura dei residui amminoacidici di superficie. Essi fanno decrescere fortemente la solubilità proteica. Questo avviene a causa della diminuzione della costante dielettrica del mezzo e grazie alla "deidratazione" (viene cioè impedita l'interazione con le molecole d'acqua). Con la diminuzione della costante dielettrica di una soluzione le forze attrattive tra residui di superficie di carica opposta aumenta e questo comporta la formazione di aggregati che precipitano. La temperatura alla quale avviene la precipitazione è un fattore importante perché in presenza di solventi organici la solubilità diminuisce marcatamente insieme alla temperatura. Comunque non bisogna dimenticarci che la precipitazione non è solo un metodo di purificazione, ma nche i concentrazione. Per es. è stato usato per l’analisi del liquido cefalo rachidiano La Precipitazione Le proteine rimangono disciolte in soluzione a causa dei loro /residui amminoacidici di superficie/ carichi che interagiscono con le molecole di solvente. Se tali interazioni vengono meno, le molecole proteiche interagiscono prevalentemente tra di loro formando aggregati che precipitano. La facilità o difficoltà con la quale la proteina viene impossibilitata a interagire col solvente dipende largamente dalla natura dei residui amminoacidici di superficie. Prove ed errori da parte dello sperimentatore serviranno a determinare il comportamento specifico della proteina. Sono 5 gli agenti comunemente usati per far precipitare le proteine: 1. sali inorganici <#Frazionamento con sali> 2. pH e temperatura <#pH e temperatura> 3. solventi organici <#Solventi organici> 4. proteine basiche <#Proteine basiche> 5. polietilenglicole <#Polietilenglicole> * Frazionamento con sali* Il metodo più usato per la precipitazione è l'aggiunta di sali come il *solfato d'ammonio* o il *fosfato di potassio*. La velocità alla quale viene aggiunto il sale alla soluzione è molto importante. Per molte proteine infatti l'aggiunta deve essere graduale. Questa velocità deve essere determinata sperimentalmente. Se è l'ammonio solfato il sale che viene aggiunto e il solvente non è tamponato o lo è debolmente, allora è importante /monitorare il pH/. Quando si rende necessario un aggiustamento del pH si può aggiungere una base debole come _/idrossido d'ammonio diluito o il Tris/_. Una volta raggiunta l'equilibrazione il precipitato viene raccolto per centrifugazione. Col solfato d'ammonio si ottiene l'allontanamento fino al 75% delle proteine grezze (ho ridotto il quantitativo di proteine a 1/4 Þ ho purificato 4X) e inoltre ho concentrato fortemente il campione ; il grado della concentrazione dipende da quanta soluzione è stata usata per ridissolvere il campione. * pH e temperatura* Come descritto in "anfoliti in soluzione acquosa " le proteine possono essere cariche positivamente o negativamente a seconda se il pH viene portato sotto il loro punto isoelettrico o sopra rispettivamente. Queste forme cariche sono molto più solubili delle molecole elettricamente neutre. Di conseguenza, cambiare il pH può portare alla precipitazione di alcune molecole. La nostra proteina può precipitare o può restare in soluzione. Quanto si possa far variare il pH e quanto rapidamente deve essere determinato sperimentalmente per ciascuna proteina. Lo stesso vale per le variazioni di temperatura. * Solventi organici* Essi fanno decrescere fortemente la solubilità proteica. Questo avviene a causa della diminuzione della costante dielettrica del mezzo e grazie alla "deidratazione" (viene cioè impedita l'interazione con le molecole d'acqua). Con la diminuzione della costante dielettrica di una soluzione le forze attrattive tra residui di superficie di carica opposta aumenta e questo comporta la formazione di aggregati che precipitano. I solventi organici che si usano più spesso sono *etanolo*, *metanolo*, *acetone*. La temperatura alla quale avviene la precipitazione è un fattore importante perché in presenza di solventi organici la solubilità diminuisce marcatamente insieme alla temperatura. * Proteine basiche* Sono policationi e si legano a composti negativamente carichi neutralizzando una grande proporzione di carica che posseggono. * Polietilenglicole* E' un polielettrolita.
. Con la diminuzione della costante dielettrica di una soluzione le forze attrattive tra residui di superficie di carica opposta aumenta e questo comporta la formazione di aggregati che precipitano. La temperatura alla quale avviene la precipitazione è un fattore importante perché in presenza di solventi organici la solubilità diminuisce marcatamente insieme alla temperatura. Comunque non bisogna dimenticarci che la precipitazione non è solo un metodo di purificazione, ma nche i concentrazione. Per es. è stato usato per l’analisi del liquido cefalo rachidiano. La Precipitazione Le proteine rimangono disciolte in soluzione a causa dei loro /residui amminoacidici di superficie/ carichi che interagiscono con le molecole di solvente. Se tali interazioni vengono meno, le molecole proteiche interagiscono prevalentemente tra di loro formando aggregati che precipitano. La facilità o difficoltà con la quale la proteina viene impossibilitata a interagire col solvente dipende largamente dalla natura dei residui amminoacidici di superficie. Prove ed errori da parte dello sperimentatore serviranno a determinare il comportamento specifico della proteina. Sono 5 gli agenti comunemente usati per far precipitare le proteine: 1. sali inorganici <#Frazionamento con sali> 2. pH e temperatura <#pH e temperatura> 3. solventi organici <#Solventi organici> 4. proteine basiche <#Proteine basiche> 5. polietilenglicole <#Polietilenglicole> * Frazionamento con sali* Il metodo più usato per la precipitazione è l aggiunta di sali come il *solfato d ammonio* o il *fosfato di potassio*. La velocità alla quale viene aggiunto il sale alla soluzione è molto importante. Per molte proteine infatti l aggiunta deve essere graduale. Questa velocità deve essere determinata sperimentalmente. Se è l ammonio solfato il sale che viene aggiunto e il solvente non è tamponato o lo è debolmente, allora è importante /monitorare il pH/. Quando si rende necessario un aggiustamento del pH si può aggiungere una base debole come _/idrossido d ammonio diluito o il Tris/_. Una volta raggiunta l equilibrazione il precipitato viene raccolto per centrifugazione. Col solfato d ammonio si ottiene l allontanamento fino al 75% delle proteine grezze (ho ridotto il quantitativo di proteine a 1/4 Þ ho purificato 4X) e inoltre ho concentrato fortemente il campione ; il grado della concentrazione dipende da quanta soluzione è stata usata per ridissolvere il campione. * pH e temperatura* Come descritto in anfoliti in soluzione acquosa le proteine possono essere cariche positivamente o negativamente a seconda se il pH viene portato sotto il loro punto isoelettrico o sopra rispettivamente. Queste forme cariche sono molto più solubili delle molecole elettricamente neutre. Di conseguenza, cambiare il pH può portare alla precipitazione di alcune molecole. La nostra proteina può precipitare o può restare in soluzione. Quanto si possa far variare il pH e quanto rapidamente deve essere determinato sperimentalmente per ciascuna proteina. Lo stesso vale per le variazioni di temperatura. * Solventi organici* Essi fanno decrescere fortemente la solubilità proteica. Questo avviene a causa della diminuzione della costante dielettrica del mezzo e grazie alla deidratazione (viene cioè impedita l interazione con le molecole d acqua). Con la diminuzione della costante dielettrica di una soluzione le forze attrattive tra residui di superficie di carica opposta aumenta e questo comporta la formazione di aggregati che precipitano. I solventi organici che si usano più spesso sono *etanolo*, *metanolo*, *acetone*. La temperatura alla quale avviene la precipitazione è un fattore importante perché in presenza di solventi organici la solubilità diminuisce marcatamente insieme alla temperatura. * Proteine basiche* Sono policationi e si legano a composti negativamente carichi neutralizzando una grande proporzione di carica che posseggono. * Polietilenglicole* E un polielettrolita.")
31
Solventi organici: TCA metanolo acetone
PRECIPITAZIONE CON SOLVENTI ORGANICI La facilità o difficoltà con la quale la proteina viene impossibilitata a interagire col solvente dipende largamente dalla natura dei residui amminoacidici di superficie. I solventi organici fanno decrescere fortemente la solubilità proteica. Questo avviene a causa della diminuzione della costante dielettrica del mezzo e grazie alla "deidratazione" (viene cioè impedita l'interazione con le molecole d'acqua). Con la diminuzione della costante dielettrica di una soluzione le forze attrattive tra residui di superficie di carica opposta aumenta e questo comporta la formazione di aggregati che precipitano. La temperatura alla quale avviene la precipitazione è un fattore importante perché in presenza di solventi organici la solubilità diminuisce marcatamente insieme alla temperatura. MEZZO APOLARE AGGREGAZIONE Solventi organici: TCA metanolo acetone TCA bassa efficienza Acetone buona TCA in acetone bassa efficienza Metanolo-cloroformio buona solo per piccoli volumi Tributil fosfato-acetone-metanolo buona
. Con la diminuzione della costante dielettrica di una soluzione le forze attrattive tra residui di superficie di carica opposta aumenta e questo comporta la formazione di aggregati che precipitano. La temperatura alla quale avviene la precipitazione è un fattore importante perché in presenza di solventi organici la solubilità diminuisce marcatamente insieme alla temperatura. MEZZO APOLARE. AGGREGAZIONE. Solventi organici: TCA. metanolo. acetone. TCA bassa efficienza. Acetone buona. TCA in acetone bassa efficienza. Metanolo-cloroformio buona solo per piccoli volumi. Tributil fosfato-acetone-metanolo buona.")
32
CONCENTRATORI CENTRIFUGHI
Molecular Weight Cut-Off 3000 5000 10000 30000 50000 100000 Membrane filtranti con MWCO Volume in ml 0,5 2 4 6 15 Membrana da ultrafiltrazione (sezione) vista al microscopio elettronico UltrafiltrazioneMembrana da ultrafiltrazione (sezione) vista al microscopio elettronico La concentrazione di una proteina mediante ultrafiltrazione consiste nel ‘forzare’ il passaggio di una soluzione attraverso una membrana semipermeabile che viene attraversata da acqua e piccoli soluti ma non dalla proteina d’interesse. La membrana (in polisulfonato o altri polimeri sintetici) ha una struttura anisotropa: i pori finissimi ( mM) che determinano la selettività del passaggio sono presenti solo a livello della ‘pelle’ della membrana. Nel caso di sistemi tipo i tubi Centricon, il passaggio della soluzione è indotto dall’accelera-zione centrifuga. Il campione è caricato nella camera superiore dell’apparato, separata dalla camera inferiore mediante una membrana appoggiata su un supporto a rete. Il tubino viene caricato su di una centrifuga a bassa velocità e la rotazione consente il passaggio di gran parte dell’acqua e sali (l’ “ultrafiltrato”) nella camera inferiore. Nella camera superiore rimane soltanto una piccola frazione del volume iniziale (una guarnizione impermeabile sul bordo della membrana impedisce che il campione vada completamente a secco) in cui la proteina si trova molto concentrata. Basterà capovolgere l’apparato e ricentrifugarlo brevemente in posizione capovolta per riottenere tutta la proteina nella provetta di recupero. Questo sistema funziona ottimamente per concentrare piccoli volumi di soluzione (fino a qualche ml).
 vista al microscopio elettronico. UltrafiltrazioneMembrana da ultrafiltrazione (sezione) vista al microscopio elettronico. La concentrazione di una proteina mediante ultrafiltrazione consiste nel ‘forzare’ il passaggio di una soluzione attraverso una membrana semipermeabile che viene attraversata da acqua e piccoli soluti ma non dalla proteina d’interesse. La membrana (in polisulfonato o altri polimeri sintetici) ha una struttura anisotropa: i pori finissimi ( mM) che determinano la selettività del passaggio sono presenti solo a livello della ‘pelle’ della membrana. Nel caso di sistemi tipo i tubi Centricon, il passaggio della soluzione è indotto dall’accelera-zione centrifuga. Il campione è caricato nella camera superiore dell’apparato, separata dalla camera inferiore mediante una membrana appoggiata su un supporto a rete. Il tubino viene caricato su di una centrifuga a bassa velocità e la rotazione consente il passaggio di gran parte dell’acqua e sali (l’ ultrafiltrato ) nella camera inferiore. Nella camera superiore rimane soltanto una piccola frazione del volume iniziale (una guarnizione impermeabile sul bordo della membrana impedisce che il campione vada completamente a secco) in cui la proteina si trova molto concentrata. Basterà capovolgere l’apparato e ricentrifugarlo brevemente in posizione capovolta per riottenere tutta la proteina nella provetta di recupero. Questo sistema funziona ottimamente per concentrare piccoli volumi di soluzione (fino a qualche ml).")
34
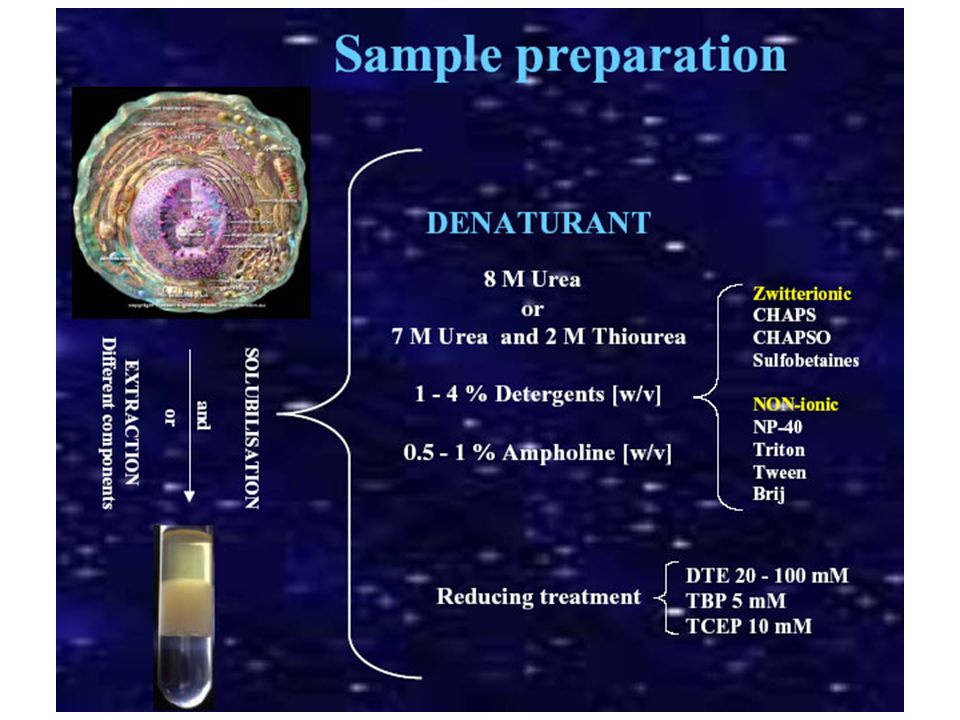
UREA: è un agente caotropico (denaturante) quindi in grado di portare ogni singola proteina ad avere una sola ed unica conformazione e quindi migrazioni omogene e di mantenere in soluzione proteine idrofobiche. Si usa a concentazioni 8-9M (alte) TIOUREA: è un agente caotropico più forte dell’Urea ed è in grado di solubilizzare meglio le proteine idrofobiche. Si usa quindi per aumentare il potere solubilizzante, ma usato in combinazione con l’UREA (7M UREA- 2M TIOUREA)]. L’azione denaturante aiuta ad inibire eventuali attività enzimatiche presenti in soluzione. Limitazione TEMPERATURA DI LAVORO Non deve superare i 30°C, altrimenti potremmo avere processi di carbamilazione L’urea è in equilibrio con l’ammonio cianato, passando per uno stadio intermedio che forma ammoniaca e acido isocianico Gli agenti caotropici rompono i legami d’idrogeno all’interno della molecola proteica La carbamilazione delle proteine porta alla modificazione dei gruppi amminici o dei gruppi sulfidirilici liberi, quindi NH2 terminale o arginine , lisine o costeine con conseguente perdita della possibilità per questi gruppi di assumere una carica positiva => si ha una alterazione del pI delle proteina i cui residui aminoacidici hanno subito tale modificazione!
 quindi in grado di portare ogni singola proteina ad avere una sola ed unica conformazione e quindi migrazioni omogene e di mantenere in soluzione proteine idrofobiche. Si usa a concentazioni 8-9M (alte) TIOUREA: è un agente caotropico più forte dell’Urea ed è in grado di solubilizzare meglio le proteine idrofobiche. Si usa quindi per aumentare il potere solubilizzante, ma usato in combinazione con l’UREA (7M UREA- 2M TIOUREA)]. L’azione denaturante aiuta ad inibire eventuali attività enzimatiche presenti in soluzione. Limitazione. TEMPERATURA DI LAVORO. Non deve superare i 30°C, altrimenti potremmo avere processi di carbamilazione. L’urea è in equilibrio con l’ammonio cianato, passando per uno stadio intermedio che forma ammoniaca e acido isocianico. Gli agenti caotropici rompono i legami d’idrogeno all’interno della molecola proteica. La carbamilazione delle proteine porta alla modificazione dei gruppi amminici o dei gruppi sulfidirilici liberi, quindi NH2 terminale o arginine , lisine o costeine con conseguente perdita della possibilità per questi gruppi di assumere una carica positiva => si ha una alterazione del pI delle proteina i cui residui aminoacidici hanno subito tale modificazione!")
35
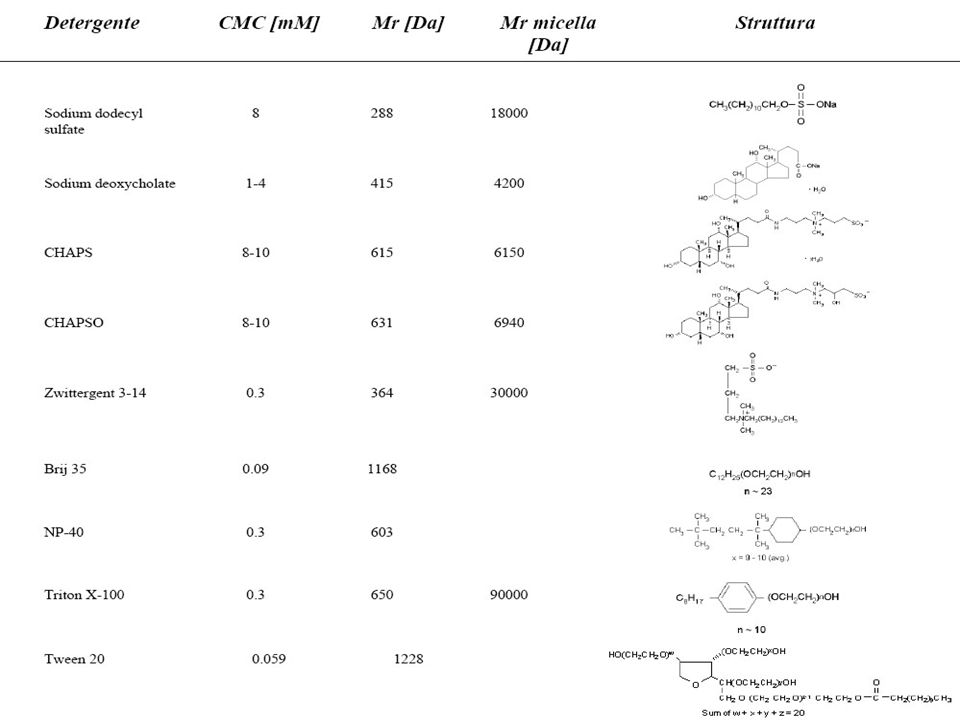
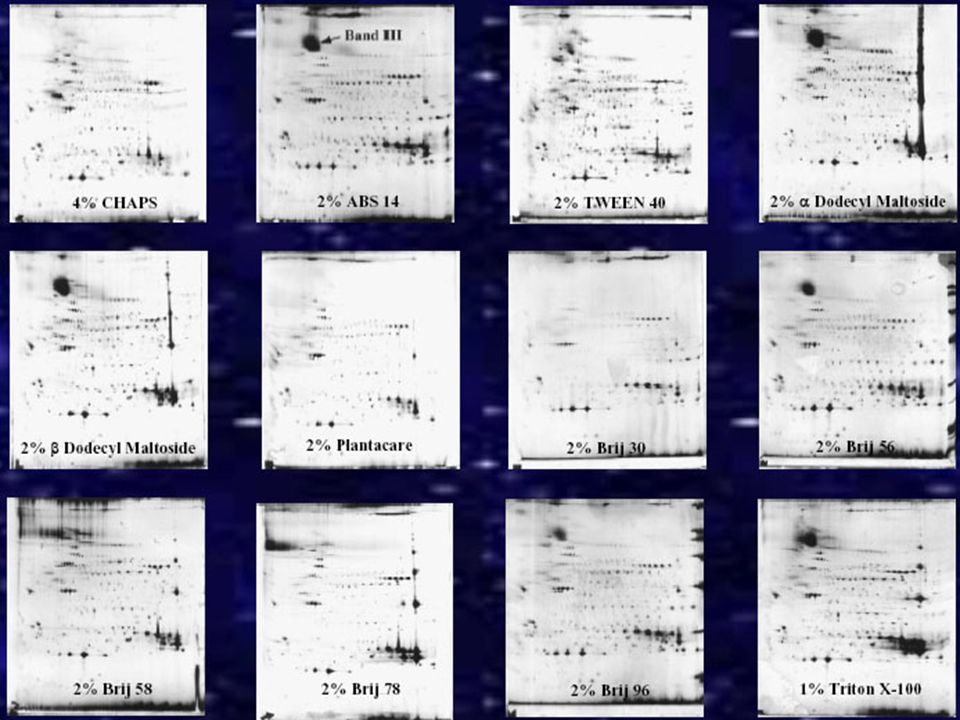
DETERGENTI Il detergente viene utilizzato per aumentare la solubilità delle proteine idrofobiche. In genere vengono utilizzati detergenti non carichi (NP-40, Triton X100, etc) o zwitterionici (CHAPS, etc). Si usano a concentrazione che variano da 1 al 4% Anche i detergenti concorrono nella denatuarzione delle proteine e nell’inibizione di attività enzimatiche presenti nel campione. Detergenti come NP-40 e Triton X100 sono non ionici e risultano essere abbastanza blandi attenzione alle attività enzimantiche che vengono mantenute (proteasi attive) SDS L’SDS essendo una molecola carica negativamente e legandosi lo stesso alle proteine (forma delle micelle detergente-proteine) non consente una corretta focalizzazione. L’SDS risulta compatibile con una isoelettrofocalizzazione qualora esso sia presente nel lisato finale ad una concentrazione minore dello 0,25% e si trovi in rapporto 1/8 (o ancora minore) con detergenti nonionici o zwitterionici I detergenti lavorano a livello delle interazioni idrofobiche e di Va
 o zwitterionici (CHAPS, etc). Si usano a concentrazione che variano da 1 al 4% Anche i detergenti concorrono nella denatuarzione delle proteine e nell’inibizione di attività enzimatiche presenti nel campione. Detergenti come NP-40 e Triton X100 sono non ionici e risultano essere abbastanza blandi attenzione alle attività enzimantiche che vengono mantenute (proteasi attive) SDS. L’SDS essendo una molecola carica negativamente e legandosi lo stesso alle proteine (forma delle micelle detergente-proteine) non consente una corretta focalizzazione. L’SDS risulta compatibile con una isoelettrofocalizzazione qualora esso sia presente nel lisato finale ad una concentrazione minore dello 0,25% e si trovi in rapporto 1/8 (o ancora minore) con detergenti nonionici o zwitterionici. I detergenti lavorano a livello delle interazioni idrofobiche e di Va.")
38
TBP a concentrazione 5mM
RIDUCENTI Agente riducente è necessario per rompere i ponti disolfuro S-S e mantenere in forma ridotta le proteina. Generalmente si usa: DTT – DiTioTreitolo o in alternativa il DTE – DiTio Eritritolo a concentrazioni mM TBP a concentrazione 5mM DTT DTE suo epimero LIMITAZIONI A pH >8 il gruppo sulfidrilico del DTT diventa ionizzato e migra perciò verso l’anodo. Questo fatto porta ad una perdita di DTT nelle regioni a pH >8 con conseguente perdita delle caratteristiche riducenti dell’ambiente. Le proteine si ossidano in modo casuale con formazione di ponti e acquisiscono carica, spostandosi dal loro punto isoelettrico (fenomeno delle strisciate di proteine a pH >8 - Streaking). Altre due strategie per evitare la formazione di ponti disolfuro sono: Ridurre ed alchilare (con iodoacetamide – IAA) la proteina prima della IEF. Ossidare i gruppi tiolici delle proteine a disulfidi misti => convertire tutti i gruppi tiolici in una forma ossidata ma stabile (HED –Hydroxyethyldisulphide – DeStreak) DeStreak
. Altre due strategie per evitare la formazione di ponti disolfuro sono: Ridurre ed alchilare (con iodoacetamide – IAA) la proteina prima della IEF. Ossidare i gruppi tiolici delle proteine a disulfidi misti => convertire tutti i gruppi tiolici in una forma ossidata ma stabile (HED –Hydroxyethyldisulphide – DeStreak) DeStreak.")
39
ANFOLINE a) piccole molecole organiche polimeriche b) altamente solubili c) anfoteriche (presenza contemporanea di gruppi basici e acidici => hanno un pI determinato da questi gruppi) d) alto potere tamponante al loro pI. Si usano nel lysis buffer per: a) aumentare la solubilità delle proteine b) prevenire interazioni proteine-gel A seconda dell’intervallo di pH scelto per l’analisi di isolettrofocusing vengono selezionate miscele di anfoliti su misura (IPG buffer - immobilzed pH gradient buffer). A: gruppo acidico B: gruppo basico
 anfoteriche (presenza contemporanea di gruppi basici e acidici => hanno un pI determinato da questi gruppi) d) alto potere tamponante al loro pI. Si usano nel lysis buffer per: a) aumentare la solubilità delle proteine. b) prevenire interazioni proteine-gel. A seconda dell’intervallo di pH scelto per l’analisi di isolettrofocusing vengono selezionate miscele di anfoliti su misura (IPG buffer - immobilzed pH gradient buffer). A: gruppo acidico. B: gruppo basico.")
40
INIBITORI DI PROTEASI:
alcuni enzimi proteolitici riescono a rimanere attivi, anche se le condizioni di preparazione sono denaturanti Per evitare una degradazione proteica durante le fasi che precedono la corsa elettroforetica, si usano: Può essere utilizzato anche Tris Base fino a 40 mM, che rende alcalino l’ambiente e neutralizza una serie di proteasi attive in ambiente acido. NUCLEASI BLUE DI BROMOFENOLO (tracce)
")
41
catene laterali polare acida non dissociabile apolare basica C O H N 2
3 acida apolare basica

42
La carica netta di una proteina dipende dal suo pI e dal pH dell’ambiente.

43
+ - - + Una proteina mostra carica elettrica = 0 ad un pH = pI
3 4 5 6 7 8 9 pH

44
verso l’anodo o verso il catodo
Elettroforesi nativa 4 5 Separazione basata sulla Carica pI = 6 In una Elettroforesi condotta a pH costante, solo quelle proteine con un pI = pH non migrano verso l’anodo o verso il catodo 8 + - - + - pH costante = 6

45
Anfoline in fase liquida
Molecole anfotere a basso peso molecolare Valori di pI leggermente spaziati con cui possiamo creare un gradiente di pH Inizialmente si sviluppavano in gel cilindrici Sottoposte ad un campo elettrico migrano a seconda della loro carica fino al pI Per la loro elevata capacità tamponante in ogni punto del gel il valore di pH sarà bene preciso Anfoline hanno valori di pI leggermente spaziati che nel loro insieme ricoprono precisi intervalli di pH. Inassenza di campo elettrico sono distribuite nel gel in maniera casuale e al momento in cui viene applicato un campo elettrico migrano verso uno dei due poli a seconda della loro carica fino a che non si fermano al valore del loro pI.

46
Limitazioni delle anfoline
Effetto del “drift catodico” Non buona riproducibilità tra esperimenti nello stesso laboratorio e tra laboratori differenti per la diversità dei lotti di anfoline Non focalizzazione delle proteine basiche Non possibilità di applicare una quantità di proteine tale da consentire successive micronalisi per il riconoscimento dei polipeptidi

47
IPG = Immobilized PH Gradient strip
Nel 1993 è stato sviluppato un metodo nel quale la focalizzazione viene svolta su gradienti IMMOBILIZZATI Il gradiente è formato legando covalentemente un gradiente di molecole cariche (immobiline) nel gel Le IMMOBILINE sono derivati della acrilammide, contenenti catene laterali con capacità tamponanti , che vengono polimerizzate nel gel insieme alla acrilammide. Il processo di formazione delle IPG è simile a quello di un gel a gradiente Nelle due camere vi sono rispettivamente un buffer acido ed uno basico La concentrazione dei buffer definisce il range di pH delle strip che si vogliono formare. Sottili strisce di gel di acrilamide montate su un supporto di plastica, che presentano un gradiente di pH creato mediante incorporazione covalente di un gradiente di tamponi nella matrice del gel nel momento in cui viene polimerizzato. Il pH in ogni punto del gel è determinato dalla miscela di molecole ‘tamponanti’ in quella posizione. Immobiline non sono altro che derivati dell’acrilammide contenenti catene laterali cariche con capacità tamponanti e che vengono polimerizzate nel gel insieme all’acrilammide. Il pH in ogni punto del gel è determinato dalla mistura di immobiline che si trovano in quel punto ed è stabile perché è immobilizzato Si versa su un supporto di plastica, si lava per allontanare i catalizzatori ed i monomeri non polimerizzati,si secca e si taglia a strip. Il pH in ogni punto del gel è determinato dalla mistura di immobiline che si trovano in quel punto.
 nel gel. Le IMMOBILINE sono derivati della acrilammide, contenenti catene laterali con capacità tamponanti , che vengono polimerizzate nel gel insieme alla acrilammide. Il processo di formazione delle IPG è simile a quello di un gel a gradiente. Nelle due camere vi sono rispettivamente un buffer acido ed uno basico. La concentrazione dei buffer definisce il range di pH delle strip che si vogliono formare. Sottili strisce di gel di acrilamide montate su un supporto di plastica, che presentano un gradiente di pH creato mediante incorporazione covalente di un gradiente di tamponi nella matrice del gel nel momento in cui viene polimerizzato. Il pH in ogni punto del gel è determinato dalla miscela di molecole ‘tamponanti’ in quella posizione. Immobiline non sono altro che derivati dell’acrilammide contenenti catene laterali cariche con capacità tamponanti e che vengono polimerizzate nel gel insieme all’acrilammide. Il pH in ogni punto del gel è determinato dalla mistura di immobiline che si trovano in quel punto ed è stabile perché è immobilizzato. Si versa su un supporto di plastica, si lava per allontanare i catalizzatori ed i monomeri non polimerizzati,si secca e si taglia a strip. Il pH in ogni punto del gel è determinato dalla mistura di immobiline che si trovano in quel punto.")
48
Vantaggi delle immobiline
Gradiente di pH stabile perché è immobilizzato Buona separazione anche a pH basici estremi Possibilità di caricare grandi quantitativi di materiale anche fino a 12 mg Riproducibilità delle mappe e confronto dei dati fra diversi laboratori

49
+ - IEF IsoElectricFocusing 4
In una elettroforesi con gradiente di pH immobilizzato, le proteine migreranno verso il rispettivo pI Proteine con 5 pI = 6 8 IEF IsoElectricFocusing + - Separazione basata sul pI 3 4 5 6 7 8 9

50
IPG strip disponibili

51
Differenze di separazione fra due gradienti
Gradiente 3-10 lineare Gradiente 3-10 Non lineare

52
Programma di focalizzazione
Reidratazione passiva Reidratazione attiva V 300V 3h V 1h 3500V 3h V 1h 9500V fino a voltaggio desiderato

53
EQUILIBRAMENTO STRIP PER LA 2° DIMENSIONE
Iodoacetamide serve per alchilare

54
- - Esse migreranno in un Gel, sotto l’azione di un campo elettrico,
+ - cariche delle catene laterali - SDS Il trattamento con SDS conferisce a tutte le proteine una carica negativa Esse migreranno in un Gel, sotto l’azione di un campo elettrico, solo in funzione della loro grandezza (M) Poliacrylamide gel Tunnel di diverso diametro Campo Elettrico Il gel viene fatto a %T lineare o a gradiente questo per aumentare la risoluzione
 Poliacrylamide gel. Tunnel di. diverso diametro. Campo Elettrico. Il gel viene fatto a %T lineare o a gradiente questo per aumentare la risoluzione.")
55
Grandezze dei pori del gel
Dipende da due grandezze: %T, %C %T= (gr. Acrilammide + gr Bis-Acrilammide/ Volume totale) x100 %C= (gr Bis-Acrilammide/ gr. Acrilammide+gr. Bis-Acrillamide) x100
 x100. %C= (gr Bis-Acrilammide/ gr. Acrilammide+gr. Bis-Acrillamide) x100.")
56
Il Gel % di acrilammide consigliata 8% 10% 12% 16%
Dimensioni delle proteine kDa kDa kDa 5-80 kDa I gel possono essere: %T costante %T a gradiente per es. 8-16%

57
4 5 6 7 8 JpH - + kD 10 20 30 40 Le strip vengono posizionate sulla sommità del gel e bloccate con una soluzione di agarosio SDS – 2D PAGE 57

58
SDS – 2D PAGE Ogni proteina (spot) è caratterizzata da Mr - pI 4 5 6 7
8 pI kD 100 50 40 Ogni proteina (spot) è caratterizzata da Mr - pI 30 20 10
 è caratterizzata. da Mr - pI")
59
Metodi per la rilevazione di spot proteici
Requisiti ideali per un metodo di rilevazione di spot proteici da 2D Alta sensibilità Permettere analisi quantitative Compatibile con MS Rapido Economico Non tossico ... in pratica non esiste il metodo ideale ....

60
COLORAZIONE DEI GEL Visibili ad occhio nudo
SILVER COOMASSIE Visibili con strumentazione RUBY DIGE

61
silver Silver acido Silver alcalino E’ il metodo più sensibile per la colorazione dei gel Sensibilità 1-2 ng Si basa sul legame che l’Ag instaura con le proteine specialmente sui gruppi amminici liberi e sulfurici SH COO- Ag+ Ag HCOH Non è reversibile in presenza di glutaraldeide Metodica lunga Colorazione non lineare se non entro certi limiti Colorazione non sempre compatibile con la massa

62
Coomassie blue E’ un metodo veloce Pratico Economico
Facilmente decolorabile Compatibile con la massa Si basa sul legame che la molecola del colorante instaura con le proteine ed esattamente con i residui di arginina e lisina La colorazione richiede un mezzo acido per la generazione di un’attrazione elettrostatica tra le molecole del colorante e i gruppi amminici delle proteine.Questa attrazione ionica, insieme con le forze di Van Der Waals mi lega il complesso colorante proteina insieme. La colorazione richiede un mezzo acido per la generaione di un’attrazione elettrostatica tra le molecole del colorante e i gruppi amminici delle proteine.Questa attrazione ionica, insieme con le forze di Van Der Waals mi lega il complesso colorante proteina insieme. La molecola del coomassie G si lega principalmente con i grupi basici dei polipeptidi (arginina e lisina) e sui residui aromatici degli ammino acidi. Quando si lega con la proteina si converte ad una forma stabile blu facilmente determinabile a 595nm Sensibilità bassa ng
 e sui residui aromatici degli ammino acidi. Quando si lega con la proteina si converte ad una forma stabile blu facilmente determinabile a 595nm. Sensibilità bassa ng.")
63
ruby E’ un metodo veloce Pratico Compatibile con la massa
Facilmente decolorabile Altamente lineare fino al 3 ordine di magnitudo Sensibilità 1-10ng Si basa sul legame che il Rubidio, un metallo chelante, instaura, in presenza di solventi organici, con i residui amminoacidici basici E’ un fluoroforo che ha due massimi di eccitazione a 280 e 450nm mentre ha un massimo di emissione a 610nm Rispetto agli altri SYPRO non c’è intercalazione con le micelle di SDS, legandosi con interazione elettrostatica con residui basici degli amminoacidi Poco economico Si deve avere una strumentazione adatta per la ripresa delle immagini

64
silver coomassie ruby

65
Strumentazione per la cattura delle immagini
VERSADOC

66
Tools del Versadoc tramite software

67
SVANTAGGI metodica lunga
bassa capacità di risoluzione delle proteine idrofobiche difficile separazione delle proteine molto acide o basiche, con valore del Punto isoelettrico estremo scarsa risoluzione di proteine con MW > 150 kDa o <10kDa bassa risoluzione delle proteine a bassa concentrazione al di sotto del 4°ordine di grandezza mancanza di linearità o sensibilità tra abbondanza della proteina nel campione proteico e capacità di risoluzione tramite colorazione sul gel

68
Identificazione Comparazione

69
Il confronto e l’analisi dei 2D Gels vengono effettuati mediante l’uso di opportuni Hardware e Software Bioinformatica 69

Presentazioni simili
















>")
1 Soluzioni e sospensioni.>")





