Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Emergenze mediche nel Diabete Mellito

2
Il Diabete Mellito Il diabete mellito è una alterazione metabolica caratterizzata da una ridotta azione dell’insulina secondaria ad un deficit dell’ormone o alla resistenza alla sua attività (o da una combinazione di questi due fattori!) Le vecchie parole "diabete mellito" descrivevano "l’eccesso di urina dolce" (GLICOSURIA) che assieme a POLIURIA e POLIDIPSIA davano la definizione classica della sindrome Caratteristica fondamentale è L’IPERGLICEMIA alla quale col tempo tendono ad associarsi le complicanze tipiche della patologia: MACROANGIOPATICHE MICROANGIOPATICHE
 Le vecchie parole diabete mellito descrivevano l’eccesso di urina dolce (GLICOSURIA) che assieme a POLIURIA e POLIDIPSIA davano la definizione classica della sindrome. Caratteristica fondamentale è L’IPERGLICEMIA alla quale col tempo tendono ad associarsi le complicanze tipiche della patologia: MACROANGIOPATICHE. MICROANGIOPATICHE.")
3
La “pandemia” diabetica

4
Classificazione una malattia tipicamente AUTO-IMMUNE
Diabete di tipo 1 una malattia tipicamente AUTO-IMMUNE il meccanismo principale è la profonda carenza insulinica interessa essenzialmente bambini e adolescenti Diabete di tipo 2 iperglicemia legata a insulino-resistenza e quindi ad una carenza insulinica relativa e progressiva. interessa essenzialmente l’adulto e rappresenta la forma di diabete PIU’ FREQUENTE comune associazione con obesità e altre malattie metaboliche

5
Altre forme di diabete mellito
Diabete secondario: Deficit Genetici della beta cellula (es. MODY 1-6) Deficit Genetici dell’azione insulinica (es. sindrome da insulino resistenza tipo A, s. di Rabson Mendenhann, lipodistrofie…) Patologie del pancreas esocrino (es. pancreatiti, pancreasectomoia, neoplasie, …) Endocrinopatie (es. acromegalia, s. di Cushing, glucagonoma, feocromocitoma, …) Jatrogeno (es. pentamidina, glucocorticoidi, clozapina, inibitori delle proteasi, acido nicotinico, ormoni tiroidei, …) Infezioni (es. rosolia congenita, CMV, coxsackie, …) Forme non comuni di DM immuno-mediato (es. “stiff-person syndrome”, recettori anti recettore insulinico…) Patologie genetiche complesse (es s. di Down, s. di Klinefelter, s. di Turner, s. di Laurence-Moon-Biedl, …) Diabete gestazionale diabete mellito sviluppato durante la gravidanza, oltre il 30 % di queste donne sviluppa il diabete di tipo 2 nel corso della vita
 Deficit Genetici dell’azione insulinica (es. sindrome da insulino resistenza tipo A, s. di Rabson Mendenhann, lipodistrofie…) Patologie del pancreas esocrino (es. pancreatiti, pancreasectomoia, neoplasie, …) Endocrinopatie (es. acromegalia, s. di Cushing, glucagonoma, feocromocitoma, …) Jatrogeno (es. pentamidina, glucocorticoidi, clozapina, inibitori delle proteasi, acido nicotinico, ormoni tiroidei, …) Infezioni (es. rosolia congenita, CMV, coxsackie, …) Forme non comuni di DM immuno-mediato (es. stiff-person syndrome , recettori anti recettore insulinico…) Patologie genetiche complesse (es s. di Down, s. di Klinefelter, s. di Turner, s. di Laurence-Moon-Biedl, …) Diabete gestazionale. diabete mellito sviluppato durante la gravidanza, oltre il 30 % di queste donne sviluppa il diabete di tipo 2 nel corso della vita.")
6
IL CONTINUUM DELLE ALTERAZIONI GLICOMETABOLICHE
Normale tolleranza ai carboidrati (NT) Glicemia a digiuno < 100 mg/dl OGTT: glicemia 2 ore dopo carico orale < 140 mg/dl Intolleranza ai carboidrati (IGT) OGTT: glicemia 2 ore dopo carico orale > 140 mg/dl e < 200 mg/dl Alterata glicemia a digiuno (IFG) Glicemia a digiuno > 100 mg/dl DIABETE MELLITO diagnosticato: In base alla glicemia Glicemia a digiuno > 126 mg/dl (riscontro confermato in almeno due occasioni!) Oppure: in presenza di sintomatologia tipica qualsiasi valore di glicemia > 200 mg/dl In base al test di carico orale con glucosio 75 g (= OGTT75 o “curva glicemica”) Glicemia 2 ore dopo carico orale > 200 mg/dl Glicemia 2 ore post- carico orale Glicemia a digiuno DM DM 200 IGT IGT= Impaired Glucose Tolerance ; IGF = Imparied Fasting Glucose 140 126 IFG 100 NT N (mg/dl) (mg/dl)
 Glicemia a digiuno < 100 mg/dl. OGTT: glicemia 2 ore dopo carico orale < 140 mg/dl. Intolleranza ai carboidrati (IGT) OGTT: glicemia 2 ore dopo carico orale > 140 mg/dl e < 200 mg/dl. Alterata glicemia a digiuno (IFG) Glicemia a digiuno > 100 mg/dl. DIABETE MELLITO diagnosticato: In base alla glicemia. Glicemia a digiuno > 126 mg/dl (riscontro confermato in almeno due occasioni!) Oppure: in presenza di sintomatologia tipica qualsiasi valore di glicemia > 200 mg/dl. In base al test di carico orale con glucosio 75 g (= OGTT75 o curva glicemica ) Glicemia 2 ore dopo carico orale > 200 mg/dl. Glicemia. 2 ore post- carico orale. Glicemia. a digiuno. DM. DM IGT. IGT= Impaired Glucose Tolerance ; IGF = Imparied Fasting Glucose IFG NT. N. (mg/dl) (mg/dl)")
8
Emergenze Metaboliche acute nel diabete mellito

9
Emergenze metaboliche
Chetoacidosi diabetica (DKA) Stato iperosmolare Acidosi Lattica Ipoglicemia Sono tutte condizioni che possono evolvere in COMA!!! Fondamentale rapida e corretta diagnosi e terapia
 Stato iperosmolare. Acidosi Lattica. Ipoglicemia. Sono tutte condizioni che possono evolvere in COMA!!! Fondamentale rapida e corretta diagnosi e terapia.")
10
KEY MESSAGE L’approccio a pazienti con una complicanza acuta critica del diabete è quello che va riservato ad ogni paziente critico!! Ricordare: Valutare pervietà vie aeree Valutare l’attività respiratoria Escludere stato di shock emodinamico La gestione dello shock viene prima di ogni indagine diagnostica Il paziente in shock ipovolemico richiede una vigorosa reidratazione con soluzione fisiologica piuttosto che supporto con vasopressori ed inotropi Altre possibili cause di shock vanno sempre ricercate soprattutto tra le emergenze addominali Posizionamento sondino-nasogastrico (specie se rischio di emesi) Il paziente in coma richiede protezione delle vie aeree (paziente in respiro spontaneocannula orofaringea; alterazioni respirointubazione orotracheale.
 Il paziente in coma richiede protezione delle vie aeree (paziente in respiro spontaneocannula orofaringea; alterazioni respirointubazione orotracheale.")
11
Chetoacidosi diabetica

12
Chetoacidosi diabetica
Sindrome caratterizzata da IPERGLICEMIA, ACIDOSI e CHETOSI, dovuta ad un deficit di insulina È l’emergenza endocrinologica più frequente (1-5 casi per ogni 100 diabetici di tipo 1/anno) Mortalità: 4-10%
 Mortalità: 4-10%")
13
Fisiopatologia L’assenza di insulina impedisce alle cellule di utilizzare il glucosio circolante per le proprie esigenze metaboliche, quindi: Accumulo di glucosio nel plasma iperglicemia marcata superamento della soglia renale di riassorbimento glicosuria poliuria sete polidipsia Utilizzo di fonti alternative di energia Riserve lipidiche perdità di massa grassa Riserve proteiche perdità di massa magra (muscolare) Nella cheto-acidosi diabetica si associano carenza insulinica e iperproduzione di glucagone dimagrimento L’eccesso di glucagone contribuisce direttamente all’iperglicemia (aumentata produzione di glucosio a livello epatico) e alla chetogensi (ossidazione degli acidi grassi liberi)
 Nella cheto-acidosi diabetica si associano carenza insulinica e iperproduzione di glucagone. dimagrimento. L’eccesso di glucagone contribuisce direttamente all’iperglicemia (aumentata produzione di glucosio a livello epatico) e alla chetogensi (ossidazione degli acidi grassi liberi)")
14
Fisiopatologia Se persiste la precedente situazione:
Disidratazione severa per poliuria ipotensione, tachicardia, torpore.. Profondo deficit energetico cellulare gli acidi grassi (forma di energia alternativa al glucosio) vengono sottoposti a b-ossidazione a livello epatico (stimolato dal glucagone) produzione di corpi chetonici (aceto-acetato acetone, acido b-idrossibutirico) chetonemia e chetonuria acidosi metabolica (cheto-acidosi) con iperventilazione secondaria e turbe digestive (vomito..) che peggiorano la disidratazione La cheto-acidosi è una complicanza spontaneamente fatale del diabete di tipo 1
 vengono sottoposti a b-ossidazione a livello epatico (stimolato dal glucagone) produzione di corpi chetonici (aceto-acetato acetone, acido b-idrossibutirico) chetonemia e chetonuria acidosi metabolica (cheto-acidosi) con iperventilazione secondaria e turbe digestive (vomito..) che peggiorano la disidratazione. La cheto-acidosi è una complicanza spontaneamente fatale del diabete di tipo 1.")
15
Riduzione dell’insulina + aumento degli ormoni contro-regolatori
lipolisi proteolisi produzione epatica del glucosio utilizzazione periferica del glucosio NEFA (sangue) aminoacidi (sangue) NEFA (epatocita) aminoacidi (epatocita) gluconeogenesi iperglicemia chetogenesi glicosuria diuresi osmotica chetosi Infezionestressaumento ormoni dello stress Qualunque sia la fonte, lo stress puo' innescare una serie di reazioni ormonali conosciuta come "risposta figth-or-flight" (letteralmente combatti o scappa). In questa reazione, il cervello manda segnali alle ghiandole surrenali costringendole a secernere ormoni quali la epinefrina (adrenalina) e norepinefrina. Questi ormoni provocano la tensione dei muscoli, l'aumento del battito cardiaco e della pressione del sangue, e un'accelerazione della respirazione. L'erogazione di sangue verso la pelle e' di gran lunga maggiore, cosi' come aumenta la sudorazione. Ogni persona che si trova a dover far fronte ad una situazione che trova stressante, come ad esempio fare un discorso di fronte ad un'ampia platea (e non si e' soliti farlo), riconoscera' questi sintomi come risposta automatica del proprio corpo a quella particolare situazione. La reazione fight-or-flight e' nata originariamente per preparare gli animali a rispondere adeguatamente a situazioni di pericolo. Si e' evoluta con il tempo, insieme all'evoluzione dell'uomo, per prepararci a rispondere fisicamente ad una minaccia (combattere o scappare). Oggigiorno, comunque, la maggior parte di noi non deve affrontare le stesse situazioni pericolose e minacciose per la propria vita come invece e' capitato ai nostri antenati. La reazione fight-or-flight non distingue piu' da una pericolo fisico e gli stress che ogni giorno la vita ci impone. In alcune persone, quando la reazione fight-or-flight si verifica ripetutamente in assenza di una minaccia reale, si puo' verificare un aumento del rischio di contrarre patologie mediche correlate allo stress. Risulta quindi importante trovare dei modi per controllare gli aspetti dannosi di questa reazione ormonale e neutralizzare gli effetti negativi che lo stress moderno ha sulla nostra salute e sul nostro benessere. L’insulina è un ormone prevalentemente “anabolico”, cioè stimolante la sintesi dei prodotti dei principali processi del metabolismo cellulare. Il suo ruolo prinicipale è quello di favorire, legandosi ad un recettore esterno della membrana cellulare, la penetrazione di glucosio nelle cellule del muscolo scheletrico, del cuore, delle ghiandole mammarie in allattamento. Stimola sintesi del gliocogeno, glicolisi, sintesi di acidi grassi e sintesi proteica. Inibisce la degradazione del glicogeno, la gluconeogenesi, la lipolisi (beta-ossidazione degli acidi grassi) e la degradazione delle preteine. Glucagone, adrenalina, cortisolo ed ormone della crescita sono ormoni controregolatori dell’azione insulinica, con attività prevalente “catabolica”, ossia stimolante la degradazione dei prodotti dei principali processi del metabolismo. Il glucagone stimola la gluconeogenesi, la degradazione del glicogeno, la lipolisi ed inibisce la sintesi del glicogeno, la glicolisi e la sintesi degli acidi grassi. L’adrenalina differisce dal glucagone in quanto il suo effetto glicogenolitico è maggiore nel muscolo. GLICOSURIA: un rene sano inizia a rilasciare glucosio nelle urine quando la concentrazione nel sangue (glicemia) supera un valore detto 'soglia renale' che corrisponde a 180 mg/dl. Pathophysiology Harrison DKA results from relative or absolute insulin deficiency combined with counterregulatory hormone excess (glucagon, catecholamines, cortisol, and growth hormone). Both insulin deficiency and glucagon excess, in particular, are necessary for DKA to develop. The decreased ratio of insulin to glucagon promotes gluconeogenesis, glycogenolysis, and ketone body formation in the liver, as well as increases in substrate delivery from fat and muscle (free fatty acids, amino acids) to the liver. The combination of insulin deficiency and hyperglycemia reduces the hepatic level of fructose-2,6-phosphate, which alters the activity of phosphofructokinase and fructose-1,6-bisphosphatase. Glucagon excess decreases the activity of pyruvate kinase, whereas insulin deficiency increases the activity of phosphoenolpyruvate carboxykinase. These changes shift the handling of pyruvate toward glucose synthesis and away from glycolysis. The increased levels of glucagon and catecholamines in the face of low insulin levels promote glycogenolysis. Insulin deficiency also reduces levels of the GLUT4 glucose transporter, which impairs glucose uptake into skeletal muscle and fat and reduces intracellular glucose metabolism (Fig ). Ketosis results from a marked increase in free fatty acid release from adipocytes, with a resulting shift toward ketone body synthesis in the liver. Reduced insulin levels, in combination with elevations in catecholamines and growth hormone, increase lipolysis and the release of free fatty acids. Normally, these free fatty acids are converted to triglycerides or VLDL in the liver. However, in DKA, hyperglucagonemia alters hepatic metabolism to favor ketone body formation, through activation of the enzyme carnitine palmitoyltransferase I. This enzyme is crucial for regulating fatty acid transport into the mitochondria, where beta oxidation and conversion to ketone bodies occur. At physiologic pH, ketone bodies exist as ketoacids, which are neutralized by bicarbonate. As bicarbonate stores are depleted, metabolic acidosis ensues. Increased lactic acid production also contributes to the acidosis. The increased free fatty acids increase triglyceride and VLDL production. VLDL clearance is also reduced because the activity of insulin-sensitive lipoprotein lipase in muscle and fat is decreased. Hypertriglyceridemia may be severe enough to cause pancreatitis. DKA is initiated by inadequate levels of plasma insulin (Table 338-5). Most commonly, DKA is precipitated by increased insulin requirements, as might occur during a concurrent illness. Failure to augment insulin therapy often compounds the problem. Occasionally, complete omission of insulin by the patient or health care team (in a hospitalized patient with type 1 DM) precipitates DKA. Patients using insulin infusion devices with short-acting insulin are at increased risk of DKA, since even a brief interruption in insulin delivery (e.g., mechanical malfunction) quickly leads to insulin deficiency. perdita degli elettroliti disidratazione cellulare riserva alcalina ipovolemia–deplez. idro-elettrolitica vomito acidosi Insufficienza Renale Funzionale ↓ escrez. chetoni e H+
 aminoacidi (sangue) NEFA (epatocita) aminoacidi (epatocita) gluconeogenesi. iperglicemia. chetogenesi. glicosuria. diuresi osmotica. chetosi. Infezionestressaumento ormoni dello stress. Qualunque sia la fonte, lo stress puo innescare una serie di reazioni ormonali conosciuta come risposta figth-or-flight (letteralmente combatti o scappa). In questa reazione, il cervello manda segnali alle ghiandole surrenali costringendole a secernere ormoni quali la epinefrina (adrenalina) e norepinefrina. Questi ormoni provocano la tensione dei muscoli, l aumento del battito cardiaco e della pressione del sangue, e un accelerazione della respirazione. L erogazione di sangue verso la pelle e di gran lunga maggiore, cosi come aumenta la sudorazione. Ogni persona che si trova a dover far fronte ad una situazione che trova stressante, come ad esempio fare un discorso di fronte ad un ampia platea (e non si e soliti farlo), riconoscera questi sintomi come risposta automatica del proprio corpo a quella particolare situazione. La reazione fight-or-flight e nata originariamente per preparare gli animali a rispondere adeguatamente a situazioni di pericolo. Si e evoluta con il tempo, insieme all evoluzione dell uomo, per prepararci a rispondere fisicamente ad una minaccia (combattere o scappare). Oggigiorno, comunque, la maggior parte di noi non deve affrontare le stesse situazioni pericolose e minacciose per la propria vita come invece e capitato ai nostri antenati. La reazione fight-or-flight non distingue piu da una pericolo fisico e gli stress che ogni giorno la vita ci impone. In alcune persone, quando la reazione fight-or-flight si verifica ripetutamente in assenza di una minaccia reale, si puo verificare un aumento del rischio di contrarre patologie mediche correlate allo stress. Risulta quindi importante trovare dei modi per controllare gli aspetti dannosi di questa reazione ormonale e neutralizzare gli effetti negativi che lo stress moderno ha sulla nostra salute e sul nostro benessere. L’insulina è un ormone prevalentemente anabolico , cioè stimolante la sintesi dei prodotti dei principali processi del metabolismo cellulare. Il suo ruolo prinicipale è quello di favorire, legandosi ad un recettore esterno della membrana cellulare, la penetrazione di glucosio nelle cellule del muscolo scheletrico, del cuore, delle ghiandole mammarie in allattamento. Stimola sintesi del gliocogeno, glicolisi, sintesi di acidi grassi e sintesi proteica. Inibisce la degradazione del glicogeno, la gluconeogenesi, la lipolisi (beta-ossidazione degli acidi grassi) e la degradazione delle preteine. Glucagone, adrenalina, cortisolo ed ormone della crescita sono ormoni controregolatori dell’azione insulinica, con attività prevalente catabolica , ossia stimolante la degradazione dei prodotti dei principali processi del metabolismo. Il glucagone stimola la gluconeogenesi, la degradazione del glicogeno, la lipolisi ed inibisce la sintesi del glicogeno, la glicolisi e la sintesi degli acidi grassi. L’adrenalina differisce dal glucagone in quanto il suo effetto glicogenolitico è maggiore nel muscolo. GLICOSURIA: un rene sano inizia a rilasciare glucosio nelle urine quando la concentrazione nel sangue (glicemia) supera un valore detto soglia renale che corrisponde a 180 mg/dl. Pathophysiology Harrison. DKA results from relative or absolute insulin deficiency combined with counterregulatory hormone excess (glucagon, catecholamines, cortisol, and growth hormone). Both insulin deficiency and glucagon excess, in particular, are necessary for DKA to develop. The decreased ratio of insulin to glucagon promotes gluconeogenesis, glycogenolysis, and ketone body formation in the liver, as well as increases in substrate delivery from fat and muscle (free fatty acids, amino acids) to the liver. The combination of insulin deficiency and hyperglycemia reduces the hepatic level of fructose-2,6-phosphate, which alters the activity of phosphofructokinase and fructose-1,6-bisphosphatase. Glucagon excess decreases the activity of pyruvate kinase, whereas insulin deficiency increases the activity of phosphoenolpyruvate carboxykinase. These changes shift the handling of pyruvate toward glucose synthesis and away from glycolysis. The increased levels of glucagon and catecholamines in the face of low insulin levels promote glycogenolysis. Insulin deficiency also reduces levels of the GLUT4 glucose transporter, which impairs glucose uptake into skeletal muscle and fat and reduces intracellular glucose metabolism (Fig ). Ketosis results from a marked increase in free fatty acid release from adipocytes, with a resulting shift toward ketone body synthesis in the liver. Reduced insulin levels, in combination with elevations in catecholamines and growth hormone, increase lipolysis and the release of free fatty acids. Normally, these free fatty acids are converted to triglycerides or VLDL in the liver. However, in DKA, hyperglucagonemia alters hepatic metabolism to favor ketone body formation, through activation of the enzyme carnitine palmitoyltransferase I. This enzyme is crucial for regulating fatty acid transport into the mitochondria, where beta oxidation and conversion to ketone bodies occur. At physiologic pH, ketone bodies exist as ketoacids, which are neutralized by bicarbonate. As bicarbonate stores are depleted, metabolic acidosis ensues. Increased lactic acid production also contributes to the acidosis. The increased free fatty acids increase triglyceride and VLDL production. VLDL clearance is also reduced because the activity of insulin-sensitive lipoprotein lipase in muscle and fat is decreased. Hypertriglyceridemia may be severe enough to cause pancreatitis. DKA is initiated by inadequate levels of plasma insulin (Table 338-5). Most commonly, DKA is precipitated by increased insulin requirements, as might occur during a concurrent illness. Failure to augment insulin therapy often compounds the problem. Occasionally, complete omission of insulin by the patient or health care team (in a hospitalized patient with type 1 DM) precipitates DKA. Patients using insulin infusion devices with short-acting insulin are at increased risk of DKA, since even a brief interruption in insulin delivery (e.g., mechanical malfunction) quickly leads to insulin deficiency. perdita degli elettroliti. disidratazione cellulare. riserva alcalina. ipovolemia–deplez. idro-elettrolitica. vomito. acidosi. Insufficienza Renale Funzionale. ↓ escrez. chetoni e H+")
16
Acidi grassi glucosio Sintesi e metabolismo dei corpi chetonici
Ciclo di Krebs citrato Acidi grassi Acetil-CoA ossidazione glucosio ossalacetato glicolisi AcetoacetilSCoA -idrossi-metil glutarilSCoA + acetil-CoA POLMONI Acido -idrossibutirrico Acido acetacetico Acetone Acetil-SCoA NAD NADH2 CO2 Sangue Tessuti (ossidazione) le proporzioni 78% a favore dell’acido beta-idrossibutirrico, 20% dell’ acido acetoacetico e 2% dell’acetone sono relativamente costanti in tutti i campioni. La beta-ossidazione è una via metabolica a spirale, che consente di degradare gli acidi grassi con produzione di acetil-CoA. L'acetil-coenzima A, spesso abbreviato in acetil-CoA, è una molecola fondamentale nel metabolismo di tutti gli organismi viventi. In alcune vie metaboliche fondamentali l'acetil coenzima A occupa una posizione centrale. Il ruolo principale è permettere l'utilizzo del prodotto della glicolisi (il piruvato) nel ciclo di Krebs. Il ciclo di Krebs (anche detto ciclo degli acidi tricarbossilici o ciclo dell'acido citrico) è un ciclo metabolico di importanza fondamentale in tutte le cellule che utilizzano ossigeno nel processo della respirazione cellulare. Il ciclo di Krebs avviene nei mitocondri delle cellule eucariote e nel citoplasma delle cellule procariote. I catabolismi glucidico e lipidico (attraverso la glicolisi e la beta ossidazione), producono acetil-CoA, un gruppo acetile legato al coenzima A. L'acetil-CoA costituisce il principale substrato del ciclo. Il suo ingresso consiste in una condensazione con ossalacetato, a generare citrato. La citrato sintasi catalizza la condensazione dell'ossalacetato con acetil-CoA, ad ottenere citrato. From libro di biochimica: L’acetil-CoA formato dall’ossidazione degli acidi grassi entra nel ciclo dell’acido citrico solo se le dgradazioni dei grassi e dei carboidrati sono adeguatamente bilanciate.. Se, invece, la degradazione lipidica predomina, l’acetil-CoA nel fegato va incontro ad un diverso destino. Il motivo di questo è che l’ingresso dell’acetil-CoA nel ciclo dell’acido citrico dipende dalla disponibilità di ossalacetato per formare citrato, ma la concenrazione di ossalacetato diminuisce se i carboidrati non sono disponibili o non vengono adeguatamente utilizzati. Durante il digiuno o in caso di diabete, l’ossalaceteto viene usato per formare glucoso e non è quindi disponibile per la condensazione con l’acetil-CoA. In queste condizioni l’acetil-CoA è deviato a formare i corpi chetonici. During high rates of fatty acid oxidation, primarily in the liver, large amounts of acetyl-CoA are generated. These exceed the capacity of the TCA cycle, and one result is the synthesis of ketone bodies, or ketogenesis. The ketone bodies are acetoacetate, b-hydroxybutyrate, and acetone. Pathophysiology Harrison DKA results from relative or absolute insulin deficiency combined with counterregulatory hormone excess (glucagon, catecholamines, cortisol, and growth hormone). Both insulin deficiency and glucagon excess, in particular, are necessary for DKA to develop. The decreased ratio of insulin to glucagon promotes gluconeogenesis, glycogenolysis, and ketone body formation in the liver, as well as increases in substrate delivery from fat and muscle (free fatty acids, amino acids) to the liver. The combination of insulin deficiency and hyperglycemia reduces the hepatic level of fructose-2,6-phosphate, which alters the activity of phosphofructokinase and fructose-1,6-bisphosphatase. Glucagon excess decreases the activity of pyruvate kinase, whereas insulin deficiency increases the activity of phosphoenolpyruvate carboxykinase. These changes shift the handling of pyruvate toward glucose synthesis and away from glycolysis. The increased levels of glucagon and catecholamines in the face of low insulin levels promote glycogenolysis. Insulin deficiency also reduces levels of the GLUT4 glucose transporter, which impairs glucose uptake into skeletal muscle and fat and reduces intracellular glucose metabolism (Fig ). Ketosis results from a marked increase in free fatty acid release from adipocytes, with a resulting shift toward ketone body synthesis in the liver. Reduced insulin levels, in combination with elevations in catecholamines and growth hormone, increase lipolysis and the release of free fatty acids. Normally, these free fatty acids are converted to triglycerides or VLDL in the liver. However, in DKA, hyperglucagonemia alters hepatic metabolism to favor ketone body formation, through activation of the enzyme carnitine palmitoyltransferase I. This enzyme is crucial for regulating fatty acid transport into the mitochondria, where beta oxidation and conversion to ketone bodies occur. At physiologic pH, ketone bodies exist as ketoacids, which are neutralized by bicarbonate. As bicarbonate stores are depleted, metabolic acidosis ensues. Increased lactic acid production also contributes to the acidosis. The increased free fatty acids increase triglyceride and VLDL production. VLDL clearance is also reduced because the activity of insulin-sensitive lipoprotein lipase in muscle and fat is decreased. Hypertriglyceridemia may be severe enough to cause pancreatitis. DKA is initiated by inadequate levels of plasma insulin (Table 338-5). Most commonly, DKA is precipitated by increased insulin requirements, as might occur during a concurrent illness. Failure to augment insulin therapy often compounds the problem. Occasionally, complete omission of insulin by the patient or health care team (in a hospitalized patient with type 1 DM) precipitates DKA. Patients using insulin infusion devices with short-acting insulin are at increased risk of DKA, since even a brief interruption in insulin delivery (e.g., mechanical malfunction) quickly leads to insulin deficiency.
 le proporzioni 78% a favore dell’acido beta-idrossibutirrico, 20% dell’ acido acetoacetico e 2% dell’acetone sono relativamente costanti in tutti i campioni. La beta-ossidazione è una via metabolica a spirale, che consente di degradare gli acidi grassi con produzione di acetil-CoA. L acetil-coenzima A, spesso abbreviato in acetil-CoA, è una molecola fondamentale nel metabolismo di tutti gli organismi viventi. In alcune vie metaboliche fondamentali l acetil coenzima A occupa una posizione centrale. Il ruolo principale è permettere l utilizzo del prodotto della glicolisi (il piruvato) nel ciclo di Krebs. Il ciclo di Krebs (anche detto ciclo degli acidi tricarbossilici o ciclo dell acido citrico) è un ciclo metabolico di importanza fondamentale in tutte le cellule che utilizzano ossigeno nel processo della respirazione cellulare. Il ciclo di Krebs avviene nei mitocondri delle cellule eucariote e nel citoplasma delle cellule procariote. I catabolismi glucidico e lipidico (attraverso la glicolisi e la beta ossidazione), producono acetil-CoA, un gruppo acetile legato al coenzima A. L acetil-CoA costituisce il principale substrato del ciclo. Il suo ingresso consiste in una condensazione con ossalacetato, a generare citrato. La citrato sintasi catalizza la condensazione dell ossalacetato con acetil-CoA, ad ottenere citrato. From libro di biochimica: L’acetil-CoA formato dall’ossidazione degli acidi grassi entra nel ciclo dell’acido citrico solo se le dgradazioni dei grassi e dei carboidrati sono adeguatamente bilanciate.. Se, invece, la degradazione lipidica predomina, l’acetil-CoA nel fegato va incontro ad un diverso destino. Il motivo di questo è che l’ingresso dell’acetil-CoA nel ciclo dell’acido citrico dipende dalla disponibilità di ossalacetato per formare citrato, ma la concenrazione di ossalacetato diminuisce se i carboidrati non sono disponibili o non vengono adeguatamente utilizzati. Durante il digiuno o in caso di diabete, l’ossalaceteto viene usato per formare glucoso e non è quindi disponibile per la condensazione con l’acetil-CoA. In queste condizioni l’acetil-CoA è deviato a formare i corpi chetonici. During high rates of fatty acid oxidation, primarily in the liver, large amounts of acetyl-CoA are generated. These exceed the capacity of the TCA cycle, and one result is the synthesis of ketone bodies, or ketogenesis. The ketone bodies are acetoacetate, b-hydroxybutyrate, and acetone. Pathophysiology Harrison. DKA results from relative or absolute insulin deficiency combined with counterregulatory hormone excess (glucagon, catecholamines, cortisol, and growth hormone). Both insulin deficiency and glucagon excess, in particular, are necessary for DKA to develop. The decreased ratio of insulin to glucagon promotes gluconeogenesis, glycogenolysis, and ketone body formation in the liver, as well as increases in substrate delivery from fat and muscle (free fatty acids, amino acids) to the liver. The combination of insulin deficiency and hyperglycemia reduces the hepatic level of fructose-2,6-phosphate, which alters the activity of phosphofructokinase and fructose-1,6-bisphosphatase. Glucagon excess decreases the activity of pyruvate kinase, whereas insulin deficiency increases the activity of phosphoenolpyruvate carboxykinase. These changes shift the handling of pyruvate toward glucose synthesis and away from glycolysis. The increased levels of glucagon and catecholamines in the face of low insulin levels promote glycogenolysis. Insulin deficiency also reduces levels of the GLUT4 glucose transporter, which impairs glucose uptake into skeletal muscle and fat and reduces intracellular glucose metabolism (Fig ). Ketosis results from a marked increase in free fatty acid release from adipocytes, with a resulting shift toward ketone body synthesis in the liver. Reduced insulin levels, in combination with elevations in catecholamines and growth hormone, increase lipolysis and the release of free fatty acids. Normally, these free fatty acids are converted to triglycerides or VLDL in the liver. However, in DKA, hyperglucagonemia alters hepatic metabolism to favor ketone body formation, through activation of the enzyme carnitine palmitoyltransferase I. This enzyme is crucial for regulating fatty acid transport into the mitochondria, where beta oxidation and conversion to ketone bodies occur. At physiologic pH, ketone bodies exist as ketoacids, which are neutralized by bicarbonate. As bicarbonate stores are depleted, metabolic acidosis ensues. Increased lactic acid production also contributes to the acidosis. The increased free fatty acids increase triglyceride and VLDL production. VLDL clearance is also reduced because the activity of insulin-sensitive lipoprotein lipase in muscle and fat is decreased. Hypertriglyceridemia may be severe enough to cause pancreatitis. DKA is initiated by inadequate levels of plasma insulin (Table 338-5). Most commonly, DKA is precipitated by increased insulin requirements, as might occur during a concurrent illness. Failure to augment insulin therapy often compounds the problem. Occasionally, complete omission of insulin by the patient or health care team (in a hospitalized patient with type 1 DM) precipitates DKA. Patients using insulin infusion devices with short-acting insulin are at increased risk of DKA, since even a brief interruption in insulin delivery (e.g., mechanical malfunction) quickly leads to insulin deficiency.")
17
CHETOACIDOSI DIABETICA
IPERGLICEMIA Altre cause: Diabete mellito, sindrome iperglicemica iperosmolare, IGT CHETOSI: Chetosi alcolica Digiuno prolungato gravidanza ACIDOSI: Acidosi lattica Acidosi ipercloremica, Abuso salicilati, Uremia, Jatrogena,

18
Eventi precipitanti la chetoacidosi
CAUSE ORGANICHE CAUSE NON ORGANICHE Nuova insorgenza di T1DM Infezioni Abuso di alcol Stress emozionale Pancreatite Emorragia gastrointestinale Terapia steroidea Infusione i.v. di destrosio Interventi maggiori Omissione della terapia - volontaria - handicap Malfunzionamento degli strumenti di somministrazione - penne - microinfusori Mancanza di insulina

19
Clinica La cheto-acidosi diabetica non è brutale
Più o meno rapida comparsa di sintomi ingravescenti: Sintomi generali: poliuria, polidipsia, disidratazione (diuresi osmotica) astenia (iperglicemia), dimagrimento (chetoacidosi), crampi, aritmie cardiache (disionie), alito chetosico (odore di frutta marcia!) Sintomi digestivi: dolori addominali frequenti, nausea, vomito.. Sintomi neurologici: sonnolenza (iperosmolarità), confusione, coma Principali segni clinici: ipotensione, anomalie ECG, disfunzioni cerebrali, perdita della massa muscolare, respiro Kussmaul, ipostenia Il miglioramento della prognosi passa per una diagnosi precoce
 astenia (iperglicemia), dimagrimento (chetoacidosi), crampi, aritmie cardiache (disionie), alito chetosico (odore di frutta marcia!) Sintomi digestivi: dolori addominali frequenti, nausea, vomito.. Sintomi neurologici: sonnolenza (iperosmolarità), confusione, coma. Principali segni clinici: ipotensione, anomalie ECG, disfunzioni cerebrali, perdita della massa muscolare, respiro Kussmaul, ipostenia. Il miglioramento della prognosi passa per una diagnosi precoce.")
20
Pattern del respiro Cheyne-Stokes Respiro Normale Biot
Respiro Apneustico Kussmaul Respiro Atassico

21
Quadro metabolico della cheto-acidosi diabetica
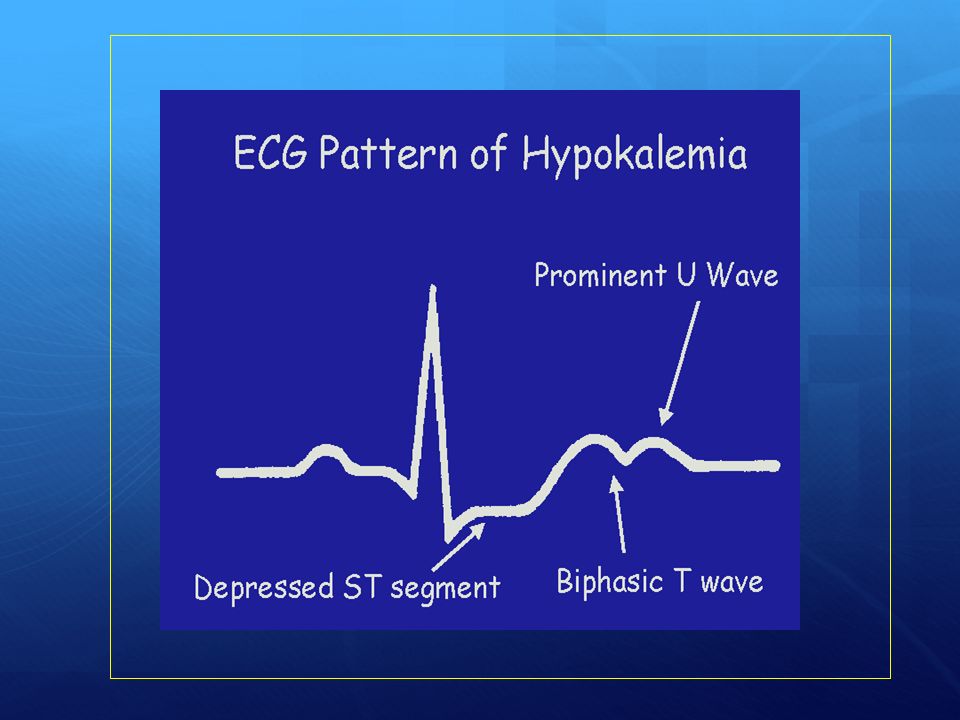
Iperglicemia severa (da 3 a 6-7 volte la norma) con aumento degli acidi grassi liberi, ipertrigliceridemia e iperosmolarità plasmatica Acidosi metabolica con diminuzione del pH e dei bicarbonati Diminuzione della pCO2 per iperventilazione di compenso Chetosi Disionie (ipokaliemia) Glicosuria e chetonuria abbondanti Disidratazione
 con aumento degli acidi grassi liberi, ipertrigliceridemia e iperosmolarità plasmatica. Acidosi metabolica con diminuzione del pH e dei bicarbonati. Diminuzione della pCO2 per iperventilazione di compenso. Chetosi. Disionie (ipokaliemia) Glicosuria e chetonuria abbondanti. Disidratazione.")
22
Tasso di mortalità della DKA nell’ultimo secolo
2000 anno

23
CHETOACIDOSI DIABETICA
CHETOSI: Altre cause: Chetosi alcolica Digiuno prolungato gravidanza ACIDOSI: Acidosi lattica Acidosi ipercloremica, Abuso salicilati, Uremia, Jatrogena, IPERGLICEMIA Diabete mellito, sindrome iperglicemica iperosmolare, IGT

24
Parametri da valutare Glicemia Chetonuria Elettroliti sierici
Glicosuria Sodio Urinocoltura Potassio Osmolarità plasmatica Magnesio Cloro Azoto Ureico Fosforo Creatinina pH Chetonemia (se disponibile) Il paziente con chetoacidosi va monitorato costantemente nei parametri vitali, laboratoristici ed ECG; spesso necessario il ricorso al CVC, SNG, catetere vescicale!
 Il paziente con chetoacidosi va monitorato costantemente nei parametri vitali, laboratoristici ed ECG; spesso necessario il ricorso al CVC, SNG, catetere vescicale!")
27
Approccio Terapeutico
Reintegrare liquidi (pz disidratato) risolvere l’acidosi e la chetosi normalizzare la glicemia Ristabilire l’equilibrio idro-elettrolitico Trattare la causa precipitante
 risolvere l’acidosi e la chetosi. normalizzare la glicemia. Ristabilire l’equilibrio idro-elettrolitico. Trattare la causa precipitante.")
28
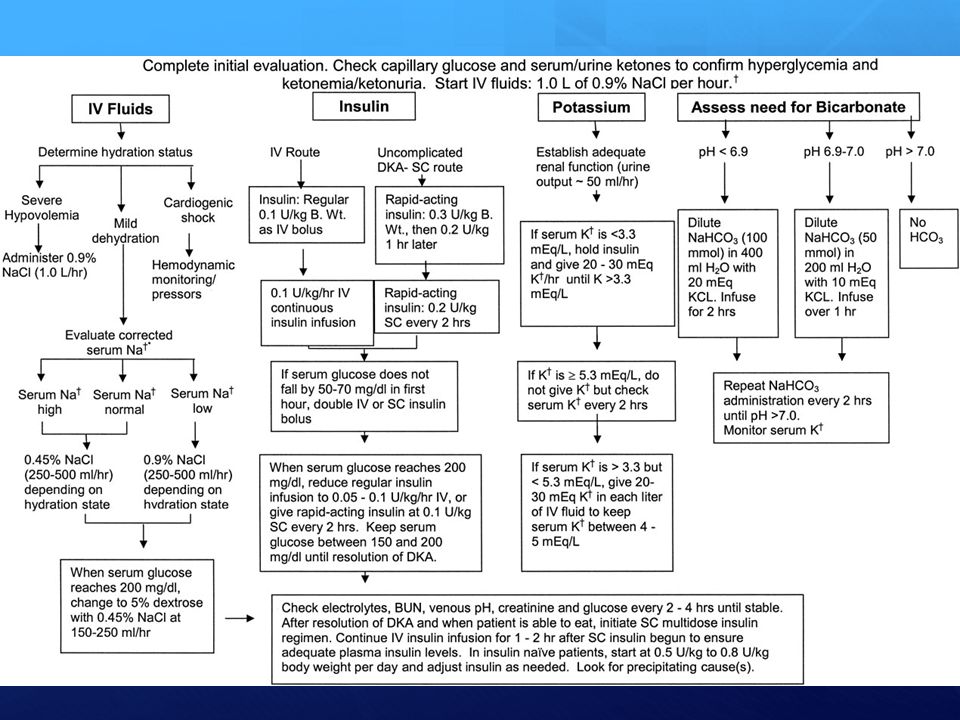
1. Reidratazione Somministrazione durante la prima ora di terapia 1000 ml di soluzione fisiologia in pazienti con normale funzionalità cardiaca (in anziani e cardiopatici iniziare con 500 ml di fisiologica e valutare la PVC) Proseguire con fisiologica ml/h (circa 4,5 ml/kg/h) nelle prime 8 ore. Se è presente ipersodiemia (Na > 155 mEq/l) si può usare salina ipotonica allo 0,45%. La velocità di flusso va regolata in base al polso,alla pressione arteriosa, alla diuresi ed al quadro neurologico. (basare la reidratazione sul valore di natriemia: riduzione oraria attesa 0,5 – 1,0 mmol/L) Una reidratazione eccessivamente aggressiva o eccessivamente ipotonica può causare edema cerebrale specie nei bambini.
 Proseguire con fisiologica ml/h (circa 4,5 ml/kg/h) nelle prime 8 ore. Se è presente ipersodiemia (Na > 155 mEq/l) si può usare salina ipotonica allo 0,45%. La velocità di flusso va regolata in base al polso,alla pressione arteriosa, alla diuresi ed al quadro neurologico. (basare la reidratazione sul valore di natriemia: riduzione oraria attesa 0,5 – 1,0 mmol/L) Una reidratazione eccessivamente aggressiva o eccessivamente. ipotonica può causare edema cerebrale specie nei bambini.")
29
2. Infusione Insulina Riduce i livelli di glucosio e previene l’ulteriore formazione di corpi chetonici, Stimola l’ingresso intracellulare del potassio già dopo minuti dalla sua somministrazione. Dose Iniziale U/Kg in bolo endovena (secondo alcuni opzionale) mantenimento: 0.1 U/Kg/h ev (all’incirca 5-7 U/h, es. 50 UI in 50 cc a 5 ml/h). Controllare le glicemie ogni ora nelle prime 4 ore. L’effetto previsto è una diminuzione della glicemia di circa 80 mg/dl ogni ora. Se la glicemia dopo la prima ora di terapia insulinica non si riduce, raddoppiare l’infusione di insulina. Una volta raggiunti livelli di glicemia inferiori a 250 mg/dl somministrare glucosata 5% per prevenire ipoglicemia.
 mantenimento: 0.1 U/Kg/h ev (all’incirca 5-7 U/h, es. 50 UI in 50 cc a 5 ml/h). Controllare le glicemie ogni ora nelle prime 4 ore. L’effetto previsto è una diminuzione della glicemia di circa 80 mg/dl ogni ora. Se la glicemia dopo la prima ora di terapia insulinica non si riduce, raddoppiare l’infusione di insulina. Una volta raggiunti livelli di glicemia inferiori a 250 mg/dl somministrare glucosata 5% per prevenire ipoglicemia.")
30
3. Correzione Disionie Potassio
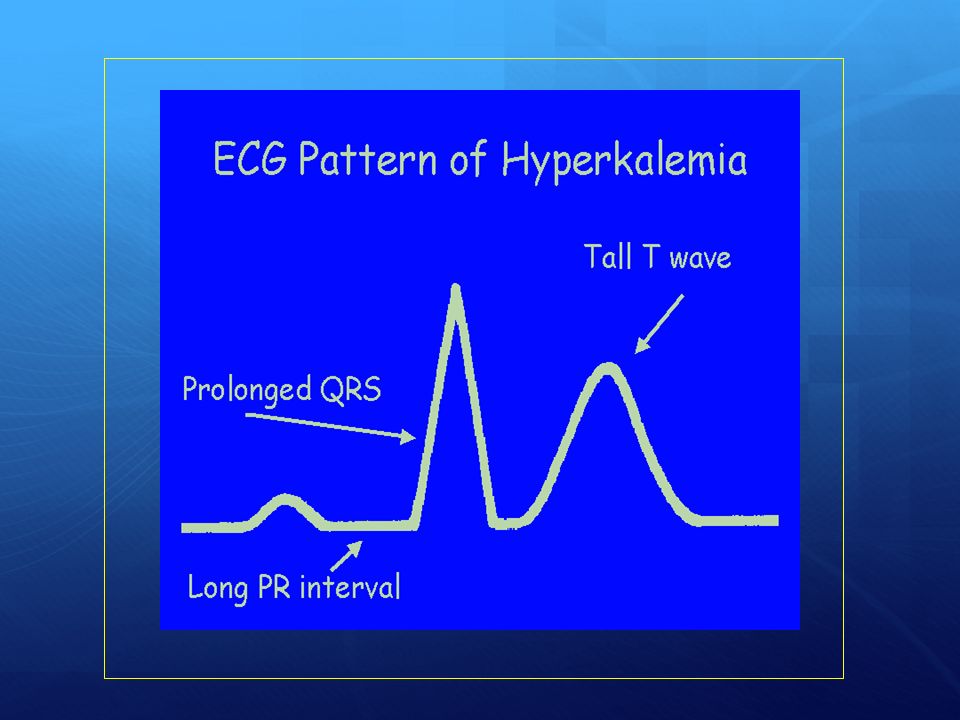
La grandezza del gradiente di potassio attraverso le membrane cellulari condiziona l’eccitabilità delle cellule nervose e muscolari (miociti compresi). Dosaggio Aggiungi mEq di potassio cloruro ogni litro di salina se il potassio plasmatico è < 5.3 mEq/l. Si può anche usare una miscela di 2/3 di KCl e 1/3 di KPO4. Controindicazioni: iperpotassiemia da insufficienza renale o da uso di farmaci potassio-risparmiatori. Precauzioni Se il potassio è inizialmente molto elevato si può procedere all’infusione di insulina contemporaneamente all’idratazione. Per valori di Potassio ematici superiori a 5.3 limitare l’infusione di potassio a 10 mEq/h NB. Per valori di kaliemia < 3,3 mmol/L non intraprendere terapia insulinica fino a reintegrazione del potassio
. Dosaggio. Aggiungi mEq di potassio cloruro ogni litro di salina se il potassio plasmatico è < 5.3 mEq/l. Si può anche usare una miscela di 2/3 di KCl e 1/3 di KPO4. Controindicazioni: iperpotassiemia da insufficienza renale o da uso di farmaci potassio-risparmiatori. Precauzioni. Se il potassio è inizialmente molto elevato si può procedere all’infusione di insulina contemporaneamente all’idratazione. Per valori di Potassio ematici superiori a 5.3 limitare l’infusione di potassio a 10 mEq/h. NB. Per valori di kaliemia < 3,3 mmol/L non intraprendere terapia insulinica fino a reintegrazione del potassio.")
31
Quando sospendere l’insulina e.v. e passare a quella s.c. ?
Risoluzione della crisi iperglicemica (glicemia < 200 mg/dL): e 2 dei seguenti criteri: bicarbonati ≥ 15 mEq/L pH venoso >7,3 gap anionico ≤ 12 mEq/L Sovrapposizione di 1-2 ore tra la somministrazione e.v. e s.c. In pz che non si alimentano per os continuare la somministrazione e.v. In pz con DM di nuova diagnosi regime terapeutico con dosi multiple di insulina pari a 0,5-0,8 U.I./Kg/die
: e 2 dei seguenti criteri: bicarbonati ≥ 15 mEq/L. pH venoso >7,3. gap anionico ≤ 12 mEq/L. Sovrapposizione di 1-2 ore tra la somministrazione e.v. e s.c. In pz che non si alimentano per os continuare la somministrazione e.v. In pz con DM di nuova diagnosi regime terapeutico con dosi multiple di insulina pari a 0,5-0,8 U.I./Kg/die.")
33
Non interrompere precocemente l’infusione di
Non cominciare l’insulina se il paziente è fortemente ipoteso, in quanto può esacerbare lo shock. Iniziare idratazione e correzione dell’ipopotassiemia prima di infondere l’insulina Non interrompere precocemente l’infusione di insulina ma aggiungere glucosio all'infusione in modo da mantenere costante la concentrazione plasmatica di insulina intorno a 100 mU/l e, quindi di conservare gli effetti massimali sull'inibizione della lipolisi e della chetogenesi.

34
Complicanze Sepsi, micosi delle mucose, infezioni polmonari e urinarie
Ischemie Trombosi Ipoglicemia tardiva Gastrite erosiva Dilatazione gastrica acuta Trombosi di catetere venoso Infarto miocardico ARDS

35
Stato iperosmolare

36
Stato Iperglicemico Iperosmolare
Iperosmolarità (osmolarità plasmatica > 320 mOsm/kg) Iperglicemia (glicemia > 600 mg/dL) di grado severo Disidratazione NON acidosi (pH sempre > 7,3 e bicarbonato > 15 mEq/L) HHS may be caused by plasma insulin concentrations that are inadequate to facilitate glucose utilization by insulinsensitive tissues but adequate (as determined by residual C-peptide) to prevent lipolysis and subsequent ketogenesis.
 Iperglicemia (glicemia > 600 mg/dL) di grado severo. Disidratazione. NON acidosi (pH sempre > 7,3 e bicarbonato > 15 mEq/L) HHS may be caused by plasma insulin concentrations that are inadequate to facilitate glucose utilization by insulinsensitive tissues but adequate (as determined by residual C-peptide) to prevent lipolysis and subsequent ketogenesis.")
37
Stato iperglicemico iperosmolare
• Deficit relativo di insulina • Aumento degli ormoni controinsulari • Eventuali perdite di liquidi (urine, tubo digerente, cute, polmone, emorragie) con disidratazione
 con disidratazione.")
38
Fattori Precipitanti Cause organiche Cause inorganiche
Infezioni (50-70%) DM di nuova insorgenza Abuso di alcol Stress emozionale Pancreatite Emorragia gastrointestinale Terapia steroidea Infusione i.v di destrosio Interventi maggiori Cause inorganiche Omissione della terapia - volontaria - handicap Involuzione cerebrale senile Perdita del senso della sete Non accessibilità fonti idriche Drugs that affect carbohydrate metabolism, such as corticosteroids, thiazides, and sympathomimetic agents (e.g., dobutamine and terbutaline) (10) and second-generation antipsychotics agents may precipitate the development of HHS or DKA. Farmaci potenzilmente responsabili di SII: Steroidi, diuretici, citotossici, difenilidantoina, diazossido, propanololo, cimetidina, fenotiazine
 DM di nuova insorgenza. Abuso di alcol. Stress emozionale. Pancreatite. Emorragia gastrointestinale. Terapia steroidea. Infusione i.v di destrosio. Interventi maggiori. Cause inorganiche. Omissione della terapia. - volontaria. - handicap. Involuzione cerebrale senile. Perdita del senso della sete. Non accessibilità fonti idriche. Drugs that affect carbohydrate metabolism, such as corticosteroids, thiazides, and sympathomimetic agents (e.g., dobutamine and terbutaline) (10) and second-generation antipsychotics agents may precipitate the development of HHS or DKA. Farmaci potenzilmente responsabili di SII: Steroidi, diuretici, citotossici, difenilidantoina, diazossido, propanololo, cimetidina, fenotiazine.")
39
Clinica Lenta evoluzione Sete intensa Poliuria
Disidratazione ingravescente Progressivo ottundimento del sensorio Tachicardia, ipotensione Shock Convulsioni Coma Esordio + lento di DKA.

40
Esami di Laboratorio Da eseguire : Potassio normale/altobasso
LAB: Glicemia, creatinina, BUN, elettroliti, es. urine, chetoni, emocromo, EGA, ev. esami colturali (urine, escreato, sangue) ed HbA1c ↑ Osmolalità [2 x (Na + K) + glicemia/18 + BUN/2.8 (v.n )] alterazioni status mentale Iperamilasemia ↑ trigliceridi STRUM.: ECG, RX torace ↑ creatinina/BUN Reperti LAB : Iperglicemia, chetosi Leucocitosi (++ neutrofili) Iposodiemia ipersodiemia pH: In CAD < 7.2; in SII > 7.3 Chetoni urinari: 4 +; glicosuria 4 + Glicemia: in CAD mg/dl; in SII anche > 1000 mg/dl Osmolalità plasmatica: 2 x (Na + K) + glicemia/18 + BUN/2.8 (v.n ): in CAD max 330 mosm/L; in SII anche > 370 mosm/L. (Studies on serum osmolality and mental alteration have established a positive linear relationship between osmolality and mental obtundation. The occurrence of stupor or coma in diabetic patients in the absence of definitive elevation of effective osmolality (320 mOsm/kg) demands immediate consideration of other causes of mental status change. In the calculation of effective osmolality {2[measured Na (mEq/l)] [glucose(mg/dl)]/18}, the urea concentration is not taken into account because it is freely permeable and its accumulation does not induce major changes in intracellular volume or osmotic gradient across the cell membrane) Chetoni: farmaci come captopril o penicillamina detrminano falsi +. Quando l’acidosi migliora, l’acido beta-OH-butirrico viene convertito in ac. Acetoacetico; i metodi di lab che utilizzano la reazione al nitroprussiato misurano solo i livelli di ac. Acetoacetico ed acetone. Quindi nelle prime fasi si potrà notare un incremento dei chetoni urinari che non riflette un peggioremento dell’acidosi e della reale chetonemia (sarebbe migliore il dosaggio sierico di acido beta-OH-butirrico) Sodiemia: nella CAD prima ridotta come conseguenza dell’iperglicemia (riduz. Na di 1.6 mEq per ogni incremento di 100 mg/dl di glicemia); poi aumenta per deplezione idrica. Nella SII, la sodiemia è dipendente dalla disidratazione ed è lievemente ridotta nei casi moderati ( mEq/l) per la vasodilatazione secondaria all’ipoglicemia (si stima una riduione di Na di 1.6 mEq per ogni incremento di 100 mg/dl di glicemia oltre i valori normali); nei casi + gravi, viceversa, raggiunge o eccede i 140 mEq/l, contribuendo quindi a sua volta in maniera significativa all’iperosmolarità. Potassio : prima aumentato (effetto dell’acidosi, simporto H+/K+), poi si riduce (effetto della tp insulinica che favorisce lo shift intracellulare di K; perdita con le urine di Sali di K; risoluzione dell’acidosi con aumento di bicarbonato che determina riduzione di K). Aumento TG pancreatite Aumento amilasi (di derivazione salivare) + dolore addominale dd. Pancreatite con lipasi Aumento BUN e creatinina: deplezione volume intravascolare + falso positivo per interferenza con acido acetoacetico
 ed HbA1c. ↑ Osmolalità [2 x (Na + K) + glicemia/18 + BUN/2.8 (v.n )] alterazioni status mentale. Iperamilasemia. ↑ trigliceridi. STRUM.: ECG, RX torace. ↑ creatinina/BUN. Reperti LAB : Iperglicemia, chetosi. Leucocitosi (++ neutrofili) Iposodiemia ipersodiemia. pH: In CAD < 7.2; in SII > 7.3. Chetoni urinari: 4 +; glicosuria 4 + Glicemia: in CAD mg/dl; in SII anche > 1000 mg/dl. Osmolalità plasmatica: 2 x (Na + K) + glicemia/18 + BUN/2.8 (v.n ): in CAD max 330 mosm/L; in SII anche > 370 mosm/L. (Studies on serum osmolality and mental alteration have established a positive linear relationship between osmolality and mental obtundation. The occurrence of stupor or coma in diabetic patients in the absence of definitive elevation of effective osmolality (320 mOsm/kg) demands immediate consideration of other causes of mental status change. In the calculation of effective osmolality {2[measured Na (mEq/l)] [glucose(mg/dl)]/18}, the urea concentration is not taken into account because it is freely permeable and its accumulation does not induce major changes in intracellular volume or osmotic gradient across the cell membrane) Chetoni: farmaci come captopril o penicillamina detrminano falsi +. Quando l’acidosi migliora, l’acido beta-OH-butirrico viene convertito in ac. Acetoacetico; i metodi di lab che utilizzano la reazione al nitroprussiato misurano solo i livelli di ac. Acetoacetico ed acetone. Quindi nelle prime fasi si potrà notare un incremento dei chetoni urinari che non riflette un peggioremento dell’acidosi e della reale chetonemia (sarebbe migliore il dosaggio sierico di acido beta-OH-butirrico) Sodiemia: nella CAD prima ridotta come conseguenza dell’iperglicemia (riduz. Na di 1.6 mEq per ogni incremento di 100 mg/dl di glicemia); poi aumenta per deplezione idrica. Nella SII, la sodiemia è dipendente dalla disidratazione ed è lievemente ridotta nei casi moderati ( mEq/l) per la vasodilatazione secondaria all’ipoglicemia (si stima una riduione di Na di 1.6 mEq per ogni incremento di 100 mg/dl di glicemia oltre i valori normali); nei casi + gravi, viceversa, raggiunge o eccede i 140 mEq/l, contribuendo quindi a sua volta in maniera significativa all’iperosmolarità. Potassio : prima aumentato (effetto dell’acidosi, simporto H+/K+), poi si riduce (effetto della tp insulinica che favorisce lo shift intracellulare di K; perdita con le urine di Sali di K; risoluzione dell’acidosi con aumento di bicarbonato che determina riduzione di K). Aumento TG pancreatite. Aumento amilasi (di derivazione salivare) + dolore addominale dd. Pancreatite con lipasi. Aumento BUN e creatinina: deplezione volume intravascolare + falso positivo per interferenza con acido acetoacetico.")
41
Approccio Terapeutico
Reintegrare liquidi (rilevanza primaria!) normalizzare la glicemia Ristabilire l’equilibrio idro-elettrolitico Trattare la causa precipitante Profilassi Tromboembolica
 normalizzare la glicemia. Ristabilire l’equilibrio idro-elettrolitico. Trattare la causa precipitante. Profilassi Tromboembolica.")
42
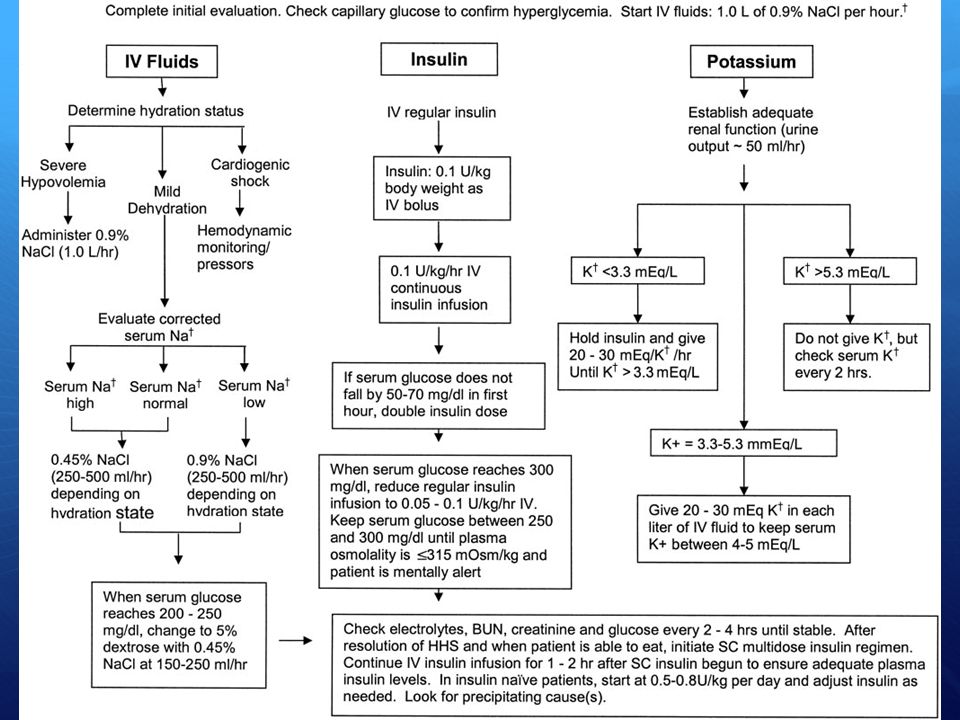
Coma/Stato iperosmolare
Glicemia >600 – Osmolarità >310 mOsm/l – pH >7.3 - Bicarbonati >14 mEq/l – Gap anionico normale <14 mEq/l Insulina Del tipo rapido a dosaggi più bassi rispetto al coma cheto-acidosico: U/kg/h Liquidi Terapia reidratante fino a 4-6 litri in 8-10 h (iniziare da 1-2 l/h, poi scendere fino a ml/h) soluzione ipotonica (NaCl 0.45%) se Na >155 soluzione fisiologica (NaCl 0.9%) se il paziente è in coma glucosata 5% 500 ml + 8 U insulina rapida se glicemia <300, per evitare ipoglicemie Può essere necessario somministrare albumina o sangue Elettroliti Sulla base delle necessità: potassio 10 mEq/h fino a controllo (controllare potassiemia, fosfatemia, ECG) Controllare equilibrio acido-base – Cateterizzare il paziente – SNG se necessario – Terapia antimicrobica se infezioni – Controllo orario glicemia
 soluzione ipotonica (NaCl 0.45%) se Na >155. soluzione fisiologica (NaCl 0.9%) se il paziente è in coma. glucosata 5% 500 ml + 8 U insulina rapida se glicemia <300, per evitare ipoglicemie. Può essere necessario somministrare albumina o sangue. Elettroliti. Sulla base delle necessità: potassio 10 mEq/h fino a controllo (controllare potassiemia, fosfatemia, ECG) Controllare equilibrio acido-base – Cateterizzare il paziente – SNG se necessario – Terapia antimicrobica se infezioni – Controllo orario glicemia.")
44
Complicanze Ipoglicemia Shock Iperglicemia EPA non cardiogeno
Ipokaliemia Ileo paralitico Acidosi metabolica ipercloremica (a gap anionico conservato) CID IRA Edema cerebrale Ipossiemia
 CID. IRA. Edema cerebrale. Ipossiemia.")
45
Ipoglicemia e coma ipoglicemico

46
Ipoglicemia Per ipoglicemia si intende una riduzione della concentrazione plasmatica di glucosio (al di sotto di mg/dl) tale da determinare l’insorgenza di sintomi che regrediscono con il ripristino dei normali livelli glicemici Tali sintomi possono comparire anche per valori superiori di glicemia qualora il calo sia eccessivamente rapido Triade di Whipple: Sintomi chiari da ipoglicemia Ridotti livelli plasmatici di glucosio (< mg/dl) Miglioramento della sintomatologia dopo tp che aumenti i livelli glicemici E’ stata sottolineata la possibilità che possano residuare deficit cognitivi permanenti (eventualità fortunatamente rara); il 2-4% delle morti da DM tipo 1 sembrerebbero imputabili ad ipoglicemia.
 tale da determinare l’insorgenza di sintomi che regrediscono con il ripristino dei normali livelli glicemici. Tali sintomi possono comparire anche per valori superiori di glicemia qualora il calo sia eccessivamente rapido. Triade di Whipple: Sintomi chiari da ipoglicemia. Ridotti livelli plasmatici di glucosio (< mg/dl) Miglioramento della sintomatologia dopo tp che aumenti i livelli glicemici. E’ stata sottolineata la possibilità che possano residuare deficit cognitivi permanenti (eventualità fortunatamente rara); il 2-4% delle morti da DM tipo 1 sembrerebbero imputabili ad ipoglicemia.")
47
Entità del Fenomeno in 45 Anni di Diabete si possono verificare:
Ipoglicemie lievi Ipoglicemie severe Coma ipoglicemico NB. Nei pz diabetici di lunga data, si ha la cosiddetta “unawareness hypoglicemia”, ipoglicemia silente o inavvertita, ossia non preceduta da sintomi adrenergici d’allarme che spingono l pz ad alimentarsi. Nei diabetici con neuropatia disautonomica si ha una ridotta risposta degli ormoni controregolatori; anche i Beta-bloccanti non selettivi possono attenuare la sintomatologia d’esordio (meno lo fanno i beta-bloccanti beta-1 selettivi). Benchè la grande maggioranza dei pz guarisca in genere dagli epidodi di ipoglicemia senza residui, è’ stata sottolineata la possibilità che possano residuare deficit cognitivi permanenti (eventualità fortunatamente rara); il 2-4% delle morti da DM tipo 1 sembrerebbero imputabili ad ipoglicemia. Diabetes, 1997 49 A
. Benchè la grande maggioranza dei pz guarisca in genere dagli epidodi di ipoglicemia senza residui, è’ stata sottolineata la possibilità che possano residuare deficit cognitivi permanenti (eventualità fortunatamente rara); il 2-4% delle morti da DM tipo 1 sembrerebbero imputabili ad ipoglicemia. Diabetes, A.")
48
Cause IPERINSULINEMIA ASSOLUTA errore di prescrizione
errore nel dosare le unità irregolarità nell’assorbimento della insulina IPERINSULINEMIA RELATIVA pasto ritardato o inadeguato attività fisica non programmata assunzione di alcolici diminuita degradazione di insulina in presenza di insufficienza epatica o renale dopo recupero da situazioni di stress al termine della gravidanza L’attività fisica aumenta l’utilizzazione di glucosio a livello muscolare e determina una riduzione del fabbisogno di insulina. Sulfaniluree: ipoglicemie gravi di durata fino a ore, soprattutto con sulf. a lunga durata d’azione (clorpropamide, gliburiide); anche sulf. a breve durata d’azione e glenidi possono però dare episodi di ipoglicemia lievi e transitori. Il rischio di ipoglicemia è > nel caso di insuff. renale o epatica, che determinano un aumento dell’emivita degli ipoglicemizzanti. Tiazolinedioni, Metformina e Acarbosio non agiscono stimolando la secrezione di insulina, e non determinano quindi ipoglicemia, ma possono però favorirla in pz trattati contemporaneamente con insulina o segretagoghi (sulfaniluree/glenidi). Ethanol blocks gluconeogenesis but not glycogenolysis. Thus, alcohol-induced hypoglycemia typically occurs after a several-day ethanol binge (bevute) during which the person eats little food, thereby causing glycogen depletion. Ethanol is usually measurable in blood at the time of presentation, but its levels correlate poorly with plasma glucose concentrations. Because gluconeogenesis becomes the predominant route of glucose production during prolonged hypoglycemia, alcohol can contribute to the progression of hypoglycemia in patients with insulin-treated diabetes.
; anche sulf. a breve durata d’azione e glenidi possono però dare episodi di ipoglicemia lievi e transitori. Il rischio di ipoglicemia è > nel caso di insuff. renale o epatica, che determinano un aumento dell’emivita degli ipoglicemizzanti. Tiazolinedioni, Metformina e Acarbosio non agiscono stimolando la secrezione di insulina, e non determinano quindi ipoglicemia, ma possono però favorirla in pz trattati contemporaneamente con insulina o segretagoghi (sulfaniluree/glenidi). Ethanol blocks gluconeogenesis but not glycogenolysis. Thus, alcohol-induced hypoglycemia typically occurs after a several-day ethanol binge (bevute) during which the person eats little food, thereby causing glycogen depletion. Ethanol is usually measurable in blood at the time of presentation, but its levels correlate poorly with plasma glucose concentrations. Because gluconeogenesis becomes the predominant route of glucose production during prolonged hypoglycemia, alcohol can contribute to the progression of hypoglycemia in patients with insulin-treated diabetes.")
49
Clinica Ansia Sonnolenza Palpitazioni Confusione mentale Tachicardia
ADRENERGICI Ansia Palpitazioni Tachicardia Tremori Sudorazione Sensazione di fame Midriasi NEUROGLICOPENICI Sonnolenza Confusione mentale Decadimento funzioni cognitive Difficoltà a parlare Incapacità a concentrarsi Spossatezza Turbe dell’umore Miosi Psicosi Convulsioni Coma I sintomi neuroglicopenici sono una diretta conseguenza della deprivazione neuronale di glucosio nel SNC. Il SNC non può utilizzare altri substrati energetici a parte il glucosio (ad es. acidi grassi), non è in grado di sintetizzare glucosio con gluconeogenesi (v. fegato) e può immagazzinare quantità di glicogeno sufficienti per fornire energia solo per pochi minuti. NB. Nei pz diabetici di lunga data, si ha la cosiddetta “unawareness hypoglicemia”, ipoglicemia silente o inavvertita, ossia non preceduta da sintomi adrenergici d’allarme che spingono l pz ad alimentarsi. Nei diabetici con neuropatia disautonomica si ha una ridotta risposta degli ormoni controregolatori; anche i Beta-bloccanti non selettivi possono attenuare la sintomatologia d’esordio (meno lo fanno i beta-bloccanti beta-1 selettivi). NON SPECIFICI Malessere Nausea Cefalea
, non è in grado di sintetizzare glucosio con gluconeogenesi (v. fegato) e può immagazzinare quantità di glicogeno sufficienti per fornire energia solo per pochi minuti. NB. Nei pz diabetici di lunga data, si ha la cosiddetta unawareness hypoglicemia , ipoglicemia silente o inavvertita, ossia non preceduta da sintomi adrenergici d’allarme che spingono l pz ad alimentarsi. Nei diabetici con neuropatia disautonomica si ha una ridotta risposta degli ormoni controregolatori; anche i Beta-bloccanti non selettivi possono attenuare la sintomatologia d’esordio (meno lo fanno i beta-bloccanti beta-1 selettivi). NON SPECIFICI. Malessere. Nausea. Cefalea.")
50
Terapia Nel paziente cosciente: somministrazione orale di zucchero (es. saccarosio g) Glucosata e.v. 20 ml al 33% poi al 10% Glucagone 1 mg i.m. o s.c. (istruire pazienti e familiari alla somministrazione del farmaco!)
")
51
Complicanze CEREBRALI Ictus CARDIACHE Aritmie
Infarto acuto del miocardio OCULARI Emorragie vitreali ALTRE Ipotermia Incidenti (sul lavoro, in auto, ecc)
")
52
Diagnosi differenziale
Coma chetoacidosico Ipoglicemia sviluppo lento, giorni veloce, minuti fame +++ sete musculatura ipotonica ipertonica, tremore cute secca umida respiro profondo, acetone normale febbre, dolori addominali delirio, ev. deficit neurologici

54
Riduzione degli Episodi Ipoglicemici Lispro vs
Riduzione degli Episodi Ipoglicemici Lispro vs. Regolare (Diabete Tipo 1) REGOLARE (n = 994) 6 7 8 9 10 LISPRO (n = 986) * Media ± 95% C.I. Episodi / 30 gg * * ULTRARAPIDA - LISPRO (Humalog® ) Ha la caratteristica di avvicinarsi molto ai tempi di reazione dell'insulina prodotta dal pancreas. Infatti la sua azione inizia dopo soli minuti dalla somministrazione per poi esaurirsi nel giro di circa 3 ore. E' una delle nuove insulina ad "azione ultrarapida“, che si differenzia dall'insulina umana "classica" perché la lisina e la prolina (due amminoacidi, appunto, e di qui il nome "lis-pro") sono state invertite sulle posizioni 28 e 29 della catena beta dell'insulina. E' proprio grazie a questa modifica chimica che la lispro è in grado di agire dopo soli minuti a fronte dei circa 30 necessari alla rapida normale. Disponibile in flaconi da 10 ml, cartucce da 3 ml e penne pre-riempite monouso da 3 ml. N.B. Le insuline ultrarapide, al contrario delle rapide o regolari che devono essere somministrate 30 minuti prima del pasto, possono essere fatte anch eappena prima del pasto. 1 2 3 * p<0.001 Mesi Anderson Jr., et al.: Diabetes 1997;46: 50 A
 REGOLARE (n = 994) LISPRO (n = 986) * Media ± 95% C.I. Episodi / 30 gg. * * ULTRARAPIDA - LISPRO (Humalog® ) Ha la caratteristica di avvicinarsi molto ai tempi di reazione dell insulina prodotta dal pancreas. Infatti la sua azione inizia dopo soli minuti dalla somministrazione per poi esaurirsi nel giro di circa 3 ore. E una delle nuove insulina ad azione ultrarapida , che si differenzia dall insulina umana classica perché la lisina e la prolina (due amminoacidi, appunto, e di qui il nome lis-pro ) sono state invertite sulle posizioni 28 e 29 della catena beta dell insulina. E proprio grazie a questa modifica chimica che la lispro è in grado di agire dopo soli minuti a fronte dei circa 30 necessari alla rapida normale. Disponibile in flaconi da 10 ml, cartucce da 3 ml e penne pre-riempite monouso da 3 ml. N.B. Le insuline ultrarapide, al contrario delle rapide o regolari che devono essere somministrate 30 minuti prima del pasto, possono essere fatte anch eappena prima del pasto * p< Mesi. Anderson Jr., et al.: Diabetes 1997;46: A.")
55
Il Profilo di Sicurezza di Lispro
Lispro riduce le ipoglicemie nel paziente di Tipo 1 Anderson et al., 1997 Lispro riduce le ipoglicemie notturne nel paziente di Tipo 2 Lispro riduce le ipoglicemie nei pazienti in terapia con CSII Zinman et al., 1997 Lispro riduce la HbA1c, senza aumentare le ipoglicemie Valle et al., 1999 Maggiore efficacia sulle ipoglicemie confermata da metanalisi Brunelle et al., 1998 57 A

56
Acidosi Lattica

57
Acidosi Lattica L’aumento di lattato (> 5 mmol/L) può essere secondario ad una aumentata produzione (ipossia) e/o ad un aumentato accumulo Il diabete per se predispone alla iperlattacidemia Nell’80-90% dei casi è secondaria ad insufficienza renale Altri fattori predisponesti sono l’insufficienza epatica, l’abuso di alcol, lo scompenso cardiaco e infezioni gravi La disponibilità di ossigeno determina la via aerobica del metabolismo glicidico con formazione di glucosio; in condizioni anaerobiche il metabolismo glicidico comporta la formazione di acido piruvico che si trasforma in acido lattico che, a sua volta, determina acidosi. L’aumento dei ivelli sierici di L-lattato può essere: Secondario a scarsa perfusione tissutale (TIPO A) da insufficienza cardiocircolatoria (IC, shock), grave anemia, deficit enzimatici mitocondriali, esposizione a inibitori (CO, cianuro) Secondario a disturbi aerobici (TIPO B) in caso di neoplasie, diabete mellito, insuff. renale o epatica, gravi infezioni (colera, malaria), convulsioni, AIDS, farmaci/tossine (biguanidi, etanolo, metanolo, isoniazide, analoghi dell’AZT e fruttosio) L’acidosi da D-lattato, comunemente associata a bypass digiuno-ileale ed a ostruzione intestinale, è dovuta a produzione di D-lattato da parte di batteri intestinali
 da insufficienza cardiocircolatoria (IC, shock), grave anemia, deficit enzimatici mitocondriali, esposizione a inibitori (CO, cianuro) Secondario a disturbi aerobici (TIPO B) in caso di neoplasie, diabete mellito, insuff. renale o epatica, gravi infezioni (colera, malaria), convulsioni, AIDS, farmaci/tossine (biguanidi, etanolo, metanolo, isoniazide, analoghi dell’AZT e fruttosio) L’acidosi da D-lattato, comunemente associata a bypass digiuno-ileale ed a ostruzione intestinale, è dovuta a produzione di D-lattato da parte di batteri intestinali.")
58
Entità del fenomeno molto rara (3 casi su abitanti), ma con alto tasso di mortalita’; dovuta ad accumulo di metformina soprattutto in pz con insufficienza renale grave (in passato fenformina): l’ incidenza puo’ essere ridotta valutando altri fattori di rischio associati: diabete non controllato, chetosi, digiuno prolungato, eccessiva assunzione di alcol, insuff. epatica e condizioni associate all’ ipossia >> il piu’ delle volte non sono state rispettate le controindicazioni
, ma con alto tasso di mortalita’; dovuta ad accumulo di metformina soprattutto in pz con insufficienza renale grave (in passato fenformina): l’ incidenza puo’ essere ridotta valutando altri fattori di rischio associati: diabete non controllato, chetosi, digiuno prolungato, eccessiva assunzione di alcol, insuff. epatica e condizioni associate all’ ipossia. >> il piu’ delle volte non sono state rispettate le controindicazioni.")
59
Attenzione agli anziani!
a causa della potenziale riduzione della funzione renale nei soggetti anziani, il dosaggio della metformina deve essere adeguato sulla base della funzione renale; e’ pertanto necessaria una valutazione periodica della funzione renale

60
Clinica Nausea Vomito Diarrea Dolori muscolari (addome e gambe)
Acidosi con iperventilazione Confusione mentale Shock Coma

61
Approccio Terapeutico
Prevenzione (prescrizione di adeguati farmaci ipoglicemizzanti) La terapia deve essere più precoce possibile e deve mirare alla reversione dello shock, della ipossia e della acidosi Una modesta alcalinizzazione è quasi sempre richiesta Bicarbonato mEq/h inizialmente e poi in base alle necessità (mantenere pH >7.2) L’utilizzazione di insulina e glucosio è di dubbia efficacia L’emodialisi è il trattamento più efficace e può essere ripetuta
 La terapia deve essere più precoce possibile e deve mirare alla reversione dello shock, della ipossia e della acidosi. Una modesta alcalinizzazione è quasi sempre richiesta. Bicarbonato mEq/h inizialmente e poi in base alle necessità (mantenere pH >7.2) L’utilizzazione di insulina e glucosio è di dubbia efficacia. L’emodialisi è il trattamento più efficace e può essere ripetuta.")
62
Caratteristiche differenziali fra i vari tipi di coma nel DM
Plasma Urine glicemia HCO3 Chetoni osmolalità glicosuria chetonuria Chetoacidosico ↑ ↓ ++++ +++ Iperosmolare = o ↓ Assenti o + +++/+ Assente o + Lattoacidosico ↑ o ↓ = Assente o + Assente o + Ipoglicemico assenti assente Assente o +

64
Chetoacidosi diabetica – Coma diabetico chetoacidosico
Glicemia >250 – pH <7.3 – Bicarbonati <15 mEq/l – Chetoni nel siero Insulina Bassi dosaggi del tipo rapido: 0.1 U/kg e.v. seguiti da 0.1 U/kg/h in infusione continua (glicemia non deve scendere di oltre 50 mg/h per rischio edema cerebrale); se glicemia non scende raddoppiare i dosaggi; se glicemia <300 dimezzare i dosaggi Nelle prime ore mantenere glicemia non <200 Continuare insulina ev fino a scomparsa di chetoni nelle urine Liquidi Vigorosa terapia reidratante con soluzione fisiologica (NaCl 0.9%): 1 l nella prima ora poi calare fino a circa 4.5 ml/kg/h (ipotonica se ipernatremia) Glucosate al 5% quando glicemia <250, per evitare ipoglicemie Elettroliti Potassio 10 mEq/h controllando potassiemia ed ECG Bicarbonato Usarli solo se pH <7.1 o K >6.5: mEq/l ipotonica, fino a riportare pH >7,2 e bicarbonati >12 mEq/l mEq di Bicarbonato da somministrare= [(P in Kg)x(0.4)x(HCO3-desiderato-HCO3misurato)]:2 in 3-4 h in soluz.glucos.5% Cateterizzare il paziente – SNG, se necessario – Terapia antimicrobica se infezioni – Controllo orario della glicemia Eparina a basso peso molecolare (per prevenzione TVP) Criteria for resolution of DKA include: glucose 200 mg/dl serum bicarbonate 18 mEq/l venous pH 7.3. Insulina: se dopo 2 h la glicemia non è scesa del 25%, si raddoppia la dose di insulina. Per evitare il rischio di edema cerebrale, non è opportuna una riduzione della glicemia di oltre mg/dl/ore Bicarbonati: secondo le LG ADA, dare solo se pH < 7.0 (50 mEql); se pH < 6.9 dare 100 mEq/l. Deficit idrico DKA: 3-8 l 8 dare 1 l la prima ora, poi 500 cc le due ore successive) Deficit idrico HHS: fino a 10 l iniziare con 1-2 l/ora per un totale di 4-6 litri in 8-10 ore
; se glicemia non scende raddoppiare i dosaggi; se glicemia <300 dimezzare i dosaggi. Nelle prime ore mantenere glicemia non <200. Continuare insulina ev fino a scomparsa di chetoni nelle urine. Liquidi. Vigorosa terapia reidratante con soluzione fisiologica (NaCl 0.9%): 1 l nella prima ora poi calare fino a circa 4.5 ml/kg/h (ipotonica se ipernatremia) Glucosate al 5% quando glicemia <250, per evitare ipoglicemie. Elettroliti. Potassio 10 mEq/h controllando potassiemia ed ECG. Bicarbonato. Usarli solo se pH <7.1 o K >6.5: mEq/l ipotonica, fino a riportare pH >7,2 e bicarbonati >12 mEq/l. mEq di Bicarbonato da somministrare= [(P in Kg)x(0.4)x(HCO3-desiderato-HCO3misurato)]:2 in 3-4 h in soluz.glucos.5% Cateterizzare il paziente – SNG, se necessario – Terapia antimicrobica se infezioni – Controllo orario della glicemia. Eparina a basso peso molecolare (per prevenzione TVP) Criteria for resolution of DKA include: glucose 200 mg/dl. serum bicarbonate 18 mEq/l. venous pH 7.3. Insulina: se dopo 2 h la glicemia non è scesa del 25%, si raddoppia la dose di insulina. Per evitare il rischio di edema cerebrale, non è opportuna una riduzione della glicemia di oltre mg/dl/ore. Bicarbonati: secondo le LG ADA, dare solo se pH < 7.0 (50 mEql); se pH < 6.9 dare 100 mEq/l. Deficit idrico DKA: 3-8 l 8 dare 1 l la prima ora, poi 500 cc le due ore successive) Deficit idrico HHS: fino a 10 l iniziare con 1-2 l/ora per un totale di 4-6 litri in 8-10 ore.")
65
Coma ipoglicemico iatrogeno (da antidiabetici o insulina)
Tavolette di zucchero, liquidi ricchi di glucosio, dolci Glucosata e.v. 20 ml al 33% poi al 10% Glucagone 1 mg i.m. o s.c. Glucagone: pz in coma. Nei pz con DM tipo 2 con ipoglicemia da sulfaniluree, non deve essere usato glucagone poiché stimola ulteriormente la secrezione di insulina (utilizzare eventualmente diazossido od octrotide). Succhi di frutta, caramelle, latte, crackers, bevande dolci. In caso di edema cerebrale mannitolo o desametasone
. Succhi di frutta, caramelle, latte, crackers, bevande dolci. In caso di edema cerebrale mannitolo o desametasone.")
66
Coma da acidosi lattica
pH <7.3 – Bicarbonati <15 mEq/l – Gap anionico >15 mEq/l Lattato sierico >5 mmol/l – Chetoni assenti Può essere provocato dalle biguanidi (fenformina) Mortalità elevata Ristabilire la perfusione tissutale Dopamina Se shock Trattamento della ipossia Alcalinizzanti Bicarbonato mEq/h inizialmente e poi in base alle necessità (mantenere pH >7.2) Emodialisi Può essere utile se bicarbonato mal tollerato
 Mortalità elevata. Ristabilire la perfusione tissutale. Dopamina. Se shock. Trattamento della ipossia. Alcalinizzanti. Bicarbonato mEq/h inizialmente e poi in base alle necessità (mantenere pH >7.2) Emodialisi. Può essere utile se bicarbonato mal tollerato.")
67
Somministrazione di potassio in pazienti con chetoacidosi diabetica
Concentrazione K Dose da somministrare < 3 mEq/l mEq/kg/h > no Non somministrare insulina prima di avere reintegrato il potassio. Se i livelli iniziali di K sono < di mmoli/l, si deve reintegrare potassio con liquidi e non si deve somministrare insulina finchè i valori di K non superano i 3.3 mmoli/l.

Presentazioni simili













. Esistono.>")








