Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
l’analisi dei processi che ne regolano:
FARMACOCINETICA E’ la branca della farmacologia che studia l’evoluzione temporale delle concentrazioni di un farmaco e dei suoi metaboliti nei diversi fluidi e tessuti dell’organismo mediante l’analisi dei processi che ne regolano: ASSORBIMENTO DISTRIBUZIONE METABOLISMO ELIMINAZIONE ADME

2
OBIETTIVI: Sviluppare nuovi farmaci
Selezionare la via di somministrazione Scegliere la forma farmaceutica Conoscere la capacità di accesso ad organi e tessuti Conoscere le vie metaboliche Caratterizzare i processi di eliminazione Stabilire le relazioni con la risposta farmacologica Migliorare i risultati dei trattamenti

3
TOSSICOCINETICA Descrive i processi di assorbimento, distribuzione, metabolismo ed escrezione (ADME) degli xenobiotici. L’assorbimento è il processo tramite cui lo xenobiotico penetra nell’organismo (sangue) dal sito di esposizione. La distribuzione è il processo di passaggio dal sangue ai vari organi e tessuti.
 dal sito di esposizione. La distribuzione è il processo di passaggio dal sangue ai vari organi e tessuti.")
4
Lo scopo della Tossicocinetica è la conoscenza del destino del tossico nell’organismo in modo da:
prevedere la relazione tra esposizione (dose x tempo x frequenza) e la durata ed intensità degli effetti tossici; prevenire o diminuire al massimo la sua permanenza nell’organismo limiti di esposizione; determinare modalità di esposizione negli studi tossicologici; eventualmente, trovare le modalità ottimali per antagonizzarne gli effetti tossici.
 e la durata ed intensità degli effetti tossici; prevenire o diminuire al massimo la sua permanenza nell’organismo limiti di esposizione; determinare modalità di esposizione negli studi tossicologici; eventualmente, trovare le modalità ottimali per antagonizzarne gli effetti tossici.")
5
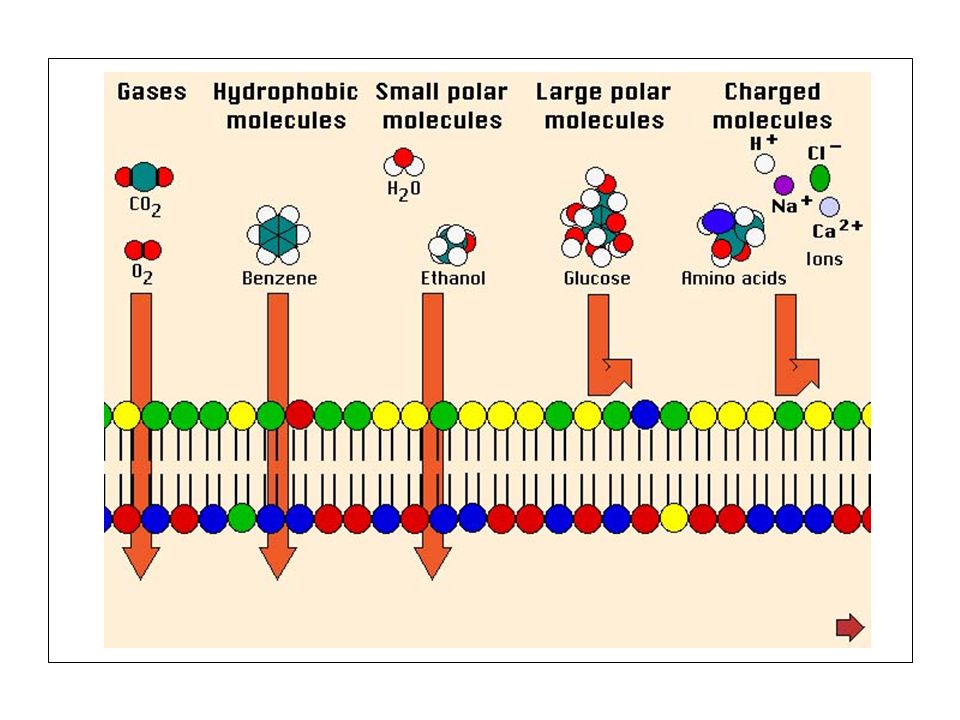
Passaggio dei farmaci attraverso le membrane cellulari
FARMACOCINETICA Studio e caratterizzazione dei fenomeni di assorbimento, distribuzione, metabolismo ed escrezione dei farmaci in funzione del tempo e dell’influenza che questi fenomeni esercitano nei confronti dell’intensità e della durata d’azione dei farmaci. Passaggio dei farmaci attraverso le membrane cellulari Membrana : mosaico di unità funzionali costituite da complessi lipoproteici Complesso lipoproteico : doppio strato di molecole fosfolipidiche perpendicolare alla superficie della membrana stessa con i gruppi polari allineati sulle due superfici della struttura e con lunghe catene idrocarboniose distese al loro interno (Dawson e Danielli).
.")
6
Teoria del modello a mosaico fluido (Singer e Nicholson):
Insieme eterogeneo di molecole globulari con i gruppi ionici ed altamente polari distribuiti sulle superfici membranali a contatto dei mezzi acquosi e con le porzioni non polari relegate nell’interno della membrana; il tutto è immerso in un doppio strato fluido e discontinuo di fosfolipidi che forma lo stroma del mosaico. Sono presenti canali per il passaggio di H2O. Lo spessore è di circa 8 nm. Le molecole dei farmaci attraversano le membrane per trasporto passivo grazie a speciali processi di trasporto. La membrana si comporta da barriera porosa lipidica inerte e il farmaco l’attraversa o diffondendo attraverso la regione lipoproteica o infiltrandosi, se la molecola è piccola, attraverso i canali idrofili.

7
La diffusibilità è direttamente proporzionale al gradiente di concentrazione esistente tra i due lati della membrana ed al coefficiente di ripartizione acqua/olio del farmaco. La maggior parte dei farmaci è costituita da acidi o basi organiche deboli in soluzione in forma ionizzata e non. Se non ionizzati, sono liposolubili, quindi in grado d’attraversare la membrana raggiungendo un equilibrio di concentrazione ai due lati di essa. Le molecole ionizzate sono spesso impossibilitate a diffondere attraverso la membrana in quanto dotate di scarsa liposolubilità. Le differenze di pH fra i due lati della membrana possono far variare il grado di ionizzazione; ad equilibrio raggiunto si avrà una più alta concentrazione del farmaco sul lato della membrana dove il grado di ionizzazione è maggiore: tale fenomeno è detto intrappolamento ionico.

8
PASSAGGIO MEDIANTE TRASPORTATORI
Acidi e basi deboli ed i loro sali possono esistere sia in forma dissociata (più idrofila) che indissociata (più lipofila). Il rapporto tra le due forme è determinato dai valori di Ka e di pH (equazione di Henderson-Hasselbach): pH = pKa + log [A-]/[AH] Nell’organismo, il pH del sangue e della maggior parte dei fluidi biologici è normalmente vicino a 7,4. Può spostarsi verso valori acidi o basici in diverse condizioni fisiologiche, patologiche o indotte da farmaci o tossici (acidosi metabolica, alcalosi respiratoria ecc.). Per sostanze con valori di pKa o pKb vicini al pH del sangue, variazioni anche lievi del pH possono far variare considerevolmente il rapporto tra specie dissociata e specie indissociata e, quindi, la capacità di attraversare le membrane. PASSAGGIO MEDIANTE TRASPORTATORI Comporta un’interazione rapidamente reversibile fra componenti costitutive della membrana e sostanze da trasferire. E’ un trasporto selettivo che dipende fondamentalmente dalla natura chimica della sostanza che transita attraverso la membrana.
 che indissociata (più lipofila). Il rapporto tra le due forme è determinato dai valori di Ka e di pH (equazione di Henderson-Hasselbach): pH = pKa + log [A-]/[AH] Nell’organismo, il pH del sangue e della maggior parte dei fluidi biologici è normalmente vicino a 7,4. Può spostarsi verso valori acidi o basici in diverse condizioni fisiologiche, patologiche o indotte da farmaci o tossici (acidosi metabolica, alcalosi respiratoria ecc.). Per sostanze con valori di pKa o pKb vicini al pH del sangue, variazioni anche lievi del pH possono far variare considerevolmente il rapporto tra specie dissociata e specie indissociata e, quindi, la capacità di attraversare le membrane. PASSAGGIO MEDIANTE TRASPORTATORI. Comporta un’interazione rapidamente reversibile fra componenti costitutive della membrana e sostanze da trasferire. E’ un trasporto selettivo che dipende fondamentalmente dalla natura chimica della sostanza che transita attraverso la membrana.")
9
E’ un processo che si giova di trasportatori ed è limitato dal fenomeno della saturazione ed è soggetto ad interferenze di competizione fra sostanze di natura chimica molto simile in concorrenza per il trasportatore stesso (inibizione competitiva). Il trasporto attivo e la diffusione facilitata sono dipendenti dall’attività di trasportatori ma il primo comporta consumo energetico assente nel secondo fenomeno (è solo aumentata la velocità del transito). Trasporto attivo: si ha nel caso di passaggio nella bile e nell’urina di farmaci fortemente acidi o alcalini Diffusione facilitata: si ha nel caso dell’ingresso del glucosio nella maggior parte delle cellule (favorite dall’insulina).
. Trasporto attivo: si ha nel caso di passaggio nella bile e nell’urina di farmaci fortemente acidi o alcalini. Diffusione facilitata: si ha nel caso dell’ingresso del glucosio nella maggior parte delle cellule (favorite dall’insulina).")
10
l’attraversano completamente.
MEMBRANA CELLULARE Proteine periferiche Proteina integrale La membrana cellulare è costituita da un doppio strato fosfolipidico le cui teste idrofile formano le superfici interna ed esterna e le code idrofobe si uniscono al centro della membrana. Il doppio strato ha uno spessore di circa 4,5 nanometri. Le proteine, che costituiscono gli altri componenti della membrana, possono essere di due tipi. Alcune dette periferiche sono disposte su entrambe le facce della membrana, altre dette integrali penetrano nella membrana e l’attraversano completamente.

11
Summary
12
Altre modalità di attraversamento delle membrane cellulari
Trasporto attivo o facilitato.

13
Proteine ed altre grosse molecole
Passaggio dei farmaci attraverso le membrane biologiche in funzione delle loro caratteristiche chimico-fisiche Caratteristiche del farmaco Passaggio attraverso le membrane biologiche PROCESSO PASSIVO Sostanze idrosolubili, non ionizzabili, con diametro molecolare inferiore a 4 Å (acqua, urea, alcool) - Filtrazione attraverso i pori Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata. Il trasferimento dipende dal pKa della sostanza e dal gradiente di pH ai due lati della membrana MECCANISMO DI TRASPORTO Sostanze idrosolubili non ionizzate con diametro superiore a 4 Å (glucosio) - Diffusione facilitata senza dispendio energetico per mezzo di un trasportatore Acidi e basi organiche ionizzate -Trasporto attivo con dispendio energetico mediante un trasportatore Proteine ed altre grosse molecole - Fagocitosi e pinocitosi (trasporto vescicolare)
 - Filtrazione attraverso i pori. Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata. Il trasferimento dipende dal pKa della sostanza e dal gradiente di pH ai due lati della membrana. MECCANISMO DI TRASPORTO. Sostanze idrosolubili non ionizzate con diametro superiore a 4 Å (glucosio) - Diffusione facilitata senza dispendio energetico per mezzo di un trasportatore. Acidi e basi organiche ionizzate. -Trasporto attivo con dispendio energetico mediante un trasportatore. Proteine ed altre grosse molecole. - Fagocitosi e pinocitosi (trasporto vescicolare)")
14
SOMMINISTRAZIONE DEI FARMACI
Per poter agire e produrre i suoi effetti sistemici, un farmaco deve prima di tutto essere assorbito e raggiungere in seguito una concentrazione efficace nel sito dove deve esercitare il suo effetto (organotropismo). La maggior parte dei farmaci viene somministrata sotto forma di medicine (preparazioni farmaceutiche e non sostanze pure); l’assorbimento del farmaco è il suo passaggio dal sito di somministrazione al torrente circolatorio; tale processo dipende dalla solubilità della preparazione, dalla via di somministrazione e dalle proprietà fisico-chimiche del composto (basso grado di ionizzazione, elevata liposolubilità della molecola).
. La maggior parte dei farmaci viene somministrata sotto forma di medicine (preparazioni farmaceutiche e non sostanze pure); l’assorbimento del farmaco è il suo passaggio dal sito di somministrazione al torrente circolatorio; tale processo dipende dalla solubilità della preparazione, dalla via di somministrazione e dalle proprietà fisico-chimiche del composto (basso grado di ionizzazione, elevata liposolubilità della molecola).")
15
Per ottenere effetti sistemici, il farmaco può essere somministrato per os o per via parenterale (iniezione o inalazione del farmaco evitando il tubo gastroenterico). Le applicazioni topiche o le infusioni intracanalicolari mammarie ed intrauterine vengono usate per ottenere effetti locali; in questi casi l’assorbimento è variabile e dipende in minima parte dalla formulazione farmaceutica oltre che dalle caratteristiche del farmaco stesso.

16
SOMMINISTRAZIONE PARENTERALE
Le principali vie parenterali sono: -IV (intravenosa) -IM (intramuscolare) -SC (sottocutanea) L’infiltrazione dei tessuti, l’intrarticolare, la subcongiuntivale e l’epidurale sono utilizzate solo per ottenere effetti localizzati. E’ importante, in tutti i casi, una rigorosa asepsi, per evitare focolai di infezione subentranti.
 -IM (intramuscolare) -SC (sottocutanea) L’infiltrazione dei tessuti, l’intrarticolare, la subcongiuntivale e l’epidurale sono utilizzate solo per ottenere effetti localizzati. E’ importante, in tutti i casi, una rigorosa asepsi, per evitare focolai di infezione subentranti.")
17
Iniezione endovenosa Prevede l’immissione di un farmaco direttamente nel torrente circolatorio, consente di ottenere una concentrazione plasmatica molto rapidamente ed in misura elevata e, spesso, un effetto farmacologico immediato. Altro vantaggio di questa via è quello di poter controllare la velocità di introduzione del farmaco in circolo. L’iniezione IV dovrebbe essere fatta sempre lentamente, tranne poche eccezioni (tiopentale, un barbiturico, somministrato rapidamente per ottenere una rapida anestesia). Talune soluzione ipertoniche o irritanti devono essere somministrate esclusivamente per questa via, mentre ne è vietato l’utilizzo nel caso di sospensioni o soluzioni oleose (vitamine liposolubili potrebbero occludere vasi di piccolo calibro con gravi effetti collaterali). Sebbene la via IV abbia molti vantaggi, è anche la più pericolosa tra le vie di somministrazione parenterali; si deve porre molta attenzione nel calcolo della dose da somministrare e nella velocità di iniezione, senza dimenticare i danni che possono essere provocati alle pareti vasali da una non corretta introduzione dell’ago.
. Talune soluzione ipertoniche o irritanti devono essere somministrate esclusivamente per questa via, mentre ne è vietato l’utilizzo nel caso di sospensioni o soluzioni oleose (vitamine liposolubili potrebbero occludere vasi di piccolo calibro con gravi effetti collaterali). Sebbene la via IV abbia molti vantaggi, è anche la più pericolosa tra le vie di somministrazione parenterali; si deve porre molta attenzione nel calcolo della dose da somministrare e nella velocità di iniezione, senza dimenticare i danni che possono essere provocati alle pareti vasali da una non corretta introduzione dell’ago.")
18
Somministrazione IM e SC
L’assorbimento dei farmaci attraverso queste vie è rapido se gli stessi sono in soluzione acquosa. Il picco di concentrazione plasmatica si manifesta entro 30 minuti. La velocità di assorbimento dipende da: -vascolarizzazione nella zona di contatto del farmaco -quantità del farmaco somministrato -sua concentrazione nella soluzione -grado di ionizzazione e di liposolubilità -area della superficie assorbente esposta al contatto del farmaco Una incompleta disponibilità sistemica può essere dovuta ad insolubilità del farmaco, al particolare pH del tessuto o a danni provocati dalla formulazione stessa nel sito di iniezione.

19
Un assorbimento rallentato attraverso la via IM è possibile ottenerlo utilizzando opportune formulazioni capaci di dare limitata disponibilità ad essere assorbite, in forza di un lento scioglimento (penicillina G procainica, ossitetraciclina base in 2-pirrolidone, protamina zinco insulina). Un assorbimento estremamente lento può essere ottenuto preparando un pellet compresso che contenga un farmaco poco o niente solubile e che sia idoneo all’impianto sottocutaneo, cosa che viene soprattutto usata per l’impiego di ormoni steroidei.

20
Assorbimento transcutaneo
Per un farmaco applicato topicamente, la possibilità di essere assorbito dalla cute dipende da due eventi conseguenti: deve sciogliersi ed essere liberato dal veicolo e deve, successivamente, penetrare nello strato cheratinizzato e nelle cellule dell’epidermide. L’assorbimento avviene per diffusione passiva, perciò è importante il grado di liposolubilità del farmaco; oltremodo importante è la concentrazione del farmaco nel veicolo (migliori quelli che contengono emulsioni olio-acqua; sostanze ad azione tensioattiva, il DMSO, le medicazioni assorbenti al polietilene per i corticosteroidi). L’ostacolo maggiore alla penetrazione transcutanea è rappresentato dallo strato corneo dell’epitelio, perciò un’area cutanea disepitelizzata consente l’assorbimento di sostanze scarsamente assorbibili dalla cute intatta. Talvolta, a seguito dell’assorbimento transcutaneo di sostanze liposolubili, possono comparire effetti tossici come accade con alcuni pesticidi ed alcuni solventi organici.
. L’ostacolo maggiore alla penetrazione transcutanea è rappresentato dallo strato corneo dell’epitelio, perciò un’area cutanea disepitelizzata consente l’assorbimento di sostanze scarsamente assorbibili dalla cute intatta. Talvolta, a seguito dell’assorbimento transcutaneo di sostanze liposolubili, possono comparire effetti tossici come accade con alcuni pesticidi ed alcuni solventi organici.")
21
Somministrazione per os
Nella maggior parte dei casi le preparazioni per os sono in forma solida: -compresse -boli -pellet -capsule anche se non mancano quelle liquide: -soluzioni acquose -elisir -sospensioni e sciroppi Prima di entrare nel circolo sistemico, il farmaco introdotto per os in forma solida subisce tre processi: -liberazione della forma farmaceutica -passaggio attraverso la mucosa gastro-enterica -transito nel parenchima epatico ed ognuno di questi processi può ridurre la quota di farmaco che entrerà nel torrente circolatorio.

22
Relazione tra concentrazione, tempo ed effetto
L’effetto di uno xenobiotico è proporzionale alla sua concentrazione nell’organo bersaglio e, in genere, al tempo di permanenza nell’organo. Dato che la concentrazione in un organo è in genere proporzionale alla concentrazione plasmatica, l’effetto è proporzionale a quest’ultima (che è misurabile). Per quasi tutti i tossici esiste una concentrazione minima, al di sotto della quale non si ha effetto.
. Per quasi tutti i tossici esiste una concentrazione minima, al di sotto della quale non si ha effetto.")
23
BIODISPONIBILITA’ Corrisponde alla velocità e alla quantità con le quali un farmaco somministrato in una certa formulazione è in grado di entrare, tal quale, nel circolo sistemico; essa può essere definita calcolando tre parametri ricavabili: -la concentrazione plasmatica picco, -il tempo necessario a conseguirla, -l’area sottesa sotto la curva costruita riunendo i valori di concentrazione determinati in tempi successivi (AUC). Il metodo più comune per calcolare la biodisponibilità sistemica (F) è dato dalla equazione : F = ( AUC ) orale ( AUC ) IV
. Il metodo più comune per calcolare la biodisponibilità sistemica (F) è dato dalla equazione : F = ( AUC ) orale. ( AUC ) IV.")
24
-un insufficiente scioglimento della preparazione (se solida)
e quando la biodisponibilità è incompleta, il rapporto delle aree è inferiore al 100 %; ciò potrebbe derivare da molteplici fattori di natura fisico/chimica e/o fisiologica quali: -un insufficiente scioglimento della preparazione (se solida) -l’instabilità o l’inattivabilità del principio attivo nel contenuto viscerale -uno scarso passaggio transmucosale. -il verificarsi di trasformazioni metaboliche nella parete intestinale o nel fegato prima dell’entrata del farmaco nel circolo sistemico. Notevoli sono le differenze di biodisponibilità di farmaci somministrati in preparazioni ad uso orale specialmente tra le specie animali monogastriche e le specie ruminanti: Nella capra (ruminante) non si ha alcun picco per l’inattivazione del principio attivo del farmaco da parte della flora microbica. Nei poligastrici è preferibile, pertanto, una via di somministrazione parenterale.
 -l’instabilità o l’inattivabilità del principio attivo nel contenuto viscerale. -uno scarso passaggio transmucosale. -il verificarsi di trasformazioni metaboliche nella parete intestinale o nel fegato prima. dell’entrata del farmaco nel circolo sistemico. Notevoli sono le differenze di biodisponibilità di farmaci somministrati in preparazioni ad uso orale specialmente tra le specie animali monogastriche e le specie ruminanti: Nella capra (ruminante) non si ha alcun picco per l’inattivazione del principio attivo del farmaco da parte della flora microbica. Nei poligastrici è preferibile, pertanto, una via di somministrazione parenterale.")
25
PURIFICAZIONE DELL’ORGANISMO
L’eliminazione totale del farmaco dall’organismo è un parametro farmacocinetico che dà un indice attendibile dell’efficacia dei meccanismi di eliminazione del farmaco, prendendo in considerazione la sommatoria dei processi esercitati da due organi emuntori (fegato, rene). Pertanto la “body clearance” (purificazione dell’organismo) è il volume di plasma ripulito da tutto il farmaco attraverso diversi processi di eliminazione nell’unità di tempo, esprimendo il parametro in termini di ml/min/kg. E’ un concetto diverso da quello di semivita in quanto permette di esprimere una entità di eliminazione di un dato farmaco indipendentemente dalla sua cinetica di disponibilità (sono comunque entrambi parametri farmacocinetici).
. Pertanto la body clearance (purificazione dell’organismo) è il volume di plasma ripulito da tutto il farmaco attraverso diversi processi di eliminazione nell’unità di tempo, esprimendo il parametro in termini di ml/min/kg. E’ un concetto diverso da quello di semivita in quanto permette di esprimere una entità di eliminazione di un dato farmaco indipendentemente dalla sua cinetica di disponibilità (sono comunque entrambi parametri farmacocinetici).")
26
La body clearance è la risultante della sommatoria dei diversi processi purificativi assicurati dai singoli organi ed è espressa come: Cl b = Cl r + Cl nr e può corrispondere per la maggior parte alla sola Cl r (clearance renale) se il farmaco è escreto esclusivamente per via renale, oppure alla Cl nr (clearance non renale) nel caso di un farmaco sottoposto ad intenso metabolismo epatico.
 se il farmaco è escreto esclusivamente per via renale, oppure alla Cl nr (clearance non renale) nel caso di un farmaco sottoposto ad intenso metabolismo epatico.")
27
Fattori che influenzano la distribuzione di un farmaco
Processo di ripartizione in tre fasi liquide: Plasma Fluidi extracellulari Fluidi intracellulari Fattori che influenzano la distribuzione di un farmaco Caratteristiche fisico-chimiche del farmaco Fissazione proteica della molecola Irrorazione degli organi Affinità specifica dei tessuti

28
DISTRIBUZIONE DEI FARMACI
I farmaci vengono trasportati all’interno dell’organismo mediante il torrente circolatorio e raggiungono i tessuti di ogni organo in misura proporzionale al flusso che giunge al singolo organo, ottenendosi, così, delle concentrazioni dipendenti dalla capacità del farmaco di attraversare l’endotelio capillare e dalla capacità di diffondere attraverso le membrane cellulari. In genere, la cinetica di distribuzione di un farmaco nel sangue, negli organi e nei tessuti è funzione: -della dose somministrata, -della via di somministrazione, -della liposolubilità del farmaco, -del grado di legame con le proteine plasmatiche.

29
Una volta nel sangue, il farmaco (libero) va incontro a due processi: distribuzione ed eliminazione. Assorbimento, distribuzione ed eliminazione avvengono contemporaneamente. Il processo di distribuzione ai tessuti è, quasi sempre, più veloce del processo di eliminazione. La velocità di distribuzione iniziale ai vari tessuti, per un dato farmaco, dipende da: 1) permeabilità capillare nei tessuti; 2) flusso ematico nel tessuto. Inizialmente si raggiungono concentrazioni elevate in organi altamente perfusi (es. fegato, reni, polmoni). La distribuzione finale dipende soprattutto dall’affinità della sostanza per i diversi tessuti. Diverse sostanze possono quindi ri-distribuirsi da tessuti ad alta perfusione a tessuti a bassa perfusione (es. tessuto adiposo, tessuto osseo) ma con più elevata affinità per il farmaco.
. La distribuzione finale dipende soprattutto dall’affinità della sostanza per i diversi tessuti. Diverse sostanze possono quindi ri-distribuirsi da tessuti ad alta perfusione a tessuti a bassa perfusione (es. tessuto adiposo, tessuto osseo) ma con più elevata affinità per il farmaco.")
30
Ad esempio, 5 minuti dopo la somministrazione di TCDD (altamente lipofila), il 15% della dose si ritrova nei polmoni e solo l’1% nel tessuto adiposo. Dopo 24 ore, il 20% della dose residua si ritrova nel tessuto adiposo, nei polmoni solo lo 0,3%. Il piombo si accumula (50% della dose) nel fegato 2 ore dopo la somministrazione. Dopo 1 mese, tuttavia, il 90% della dose residua si trova nel tessuto osseo.
 nel fegato 2 ore dopo la somministrazione. Dopo 1 mese, tuttavia, il 90% della dose residua si trova nel tessuto osseo.")
31
Velocità di eliminazione di un farmaco
E’ soprattutto in funzione dei meccanismi con cui tale processo avviene; può infatti essere influenzata da: -un massivo legame con le proteine del plasma -il grado di irrorazione degli organi emuntori -l’attività degli enzimi farmaco-metabolizzanti -l’efficacia dell’ escrezione renale. Alcune sostanze si accumulano in alcuni tessuti in conseguenza di alta liposolubilità, legame con proteine tissutali, trasporto attivo. Spesso il tessuto di deposito non è il tessuto bersaglio. In questo caso l’accumulo ha un effetto protettivo poiché diminuisce le concentrazioni nel plasma e nell’organo bersaglio. Tuttavia, il tossico viene reimmesso nel sangue man mano che la quota in circolazione viene eliminata permanenza di basse concentrazioni plasmatiche per lungo tempo.

32
TESSUTI DI DEPOSITO Proteine plasmatiche. Principalmente albumina Fegato e rene. Alcune proteine legano tossici (anioni organici, metalli pesanti) con alta affinità. La captazione avviene anche tramite sistemi di trasporto. Tessuto adiposo. Per sostanze lipofile. Tessuto osseo. Anioni e cationi (F, Pb, Sr) possono essere incorporati nella matrice ossea. Le sostanze liposolubili, compresi i gas, si distribuiscono nei compartimenti lipidici dell’organismo. Il tessuto adiposo è poco irrorato e si equilibra quindi lentamente con il sangue. Può però agire da deposito per queste sostanze, rilasciandone quantità rilevanti nel sangue dopo la fine dell’esposizione.
 con alta affinità. La captazione avviene anche tramite sistemi di trasporto. Tessuto adiposo. Per sostanze lipofile. Tessuto osseo. Anioni e cationi (F, Pb, Sr) possono essere incorporati nella matrice ossea. Le sostanze liposolubili, compresi i gas, si distribuiscono nei compartimenti lipidici dell’organismo. Il tessuto adiposo è poco irrorato e si equilibra quindi lentamente con il sangue. Può però agire da deposito per queste sostanze, rilasciandone quantità rilevanti nel sangue dopo la fine dell’esposizione.")
33
Le sostanze lipofile si distribuiscono rapidamente nel SNC, che ha un flusso ematico abbastanza elevato (350 ml/min). Il SNC, inoltre, è ricco di lipidi. Le sostanze lipofile si concentrano quindi nel SNC, oltre che nel tessuto adiposo. Le sostanze lipofile sono escrete più lentamente di quelle idrofile perché: Sono più facilmente riassorbite nel tubulo renale (o nell’intestino in caso di eliminazione biliare); si accumulano nel tessuto adiposo
; si accumulano nel tessuto adiposo.")
34
Il valore di semivita Viene identificato misurando semplicemente il tempo necessario perché ogni concentrazione plasmatica possa ridursi del 50 %. I valori di semivita della maggior parte dei farmaci utilizzati in terapia sono indipendenti dalla dose somministrata, così come anche dalla via di somministrazione, a patto che l’assorbimento del farmaco, sia esso somministrato per os o per via parenterale, sia molto rapido. La velocità di eliminazione di un farmaco è molto importante nel condizionare la durata del suo effetto farmacologico; in questa ottica, assieme con la gamma delle concentrazioni plasmatiche terapeutiche, si usa il valore di semivita per programmare gli intervalli di somministrazione di un farmaco che debba essere somministrato ripetutamente per assicurare una particolare costanza di effetto (digossina nel trattamento dell’ insufficienza cardiaca congestizia; fenitoina nella terapia anticonvulsivante).
.")
35
Fattori che interferiscono col tempo di semivita
Qualsiasi condizione fisiologica o stato patologico che alteri l’accesso di un farmaco negli organi preposti alla sua eliminazione o che comunque alteri l’ efficacia dei meccanismi eliminativi è in grado di provocare variazioni della normale semivita del farmaco. I processi biotrasformativi dovuti al sistema di enzimi microsomiali epatici, così come la funzione renale, insufficienti nei neonati, sono fattori fisiologici che allungano il tempo di semivita di farmaci che prevedono interventi metabolici a carico di tali distretti. Il pH urinario può variare la semivita di un farmaco sottoposto a riassorbimento tubulare: -se si alcalinizzano le urine, si assiste a riduzione del riassorbimento di acidi organici deboli e di conseguenza all’accorciamento della loro semivita; -il contrario accade per le basi organiche deboli con l’acidificazione delle urine. Notevolissime le differenze specie-specifiche per quanto riguarda la semivita di parecchi farmaci; dal momento che, come si è visto, notevoli sono le differenze specie-specifiche per quanto riguarda la biodisponibilità.

36
LEGAME ALLE PROTEINE PLASMATICHE
E’ un fattore che può limitare la possibilità di distribuzione, la disponibilità a raggiungere la biofase e può condizionare l’eliminabilità dall’organismo. E’ una interazione reversibile che costituisce un serbatoio di farmaco attivo. La proteina che lega la maggior parte dei farmaci è l’albumina. Il legame del farmaco all’albumina costituisce un serbatoio di farmaco inattivo. Sarà naturalmente attivo quando si stacca dalla proteina trasportatrice. Il legame farmaco-proteina riduce la possibilità di filtrazione glomerulare del farmaco mentre non interferisce con l’escrezione tubulare dipendente da specifici trasportatori. Il trasportatore specifico è in grado di eliminare il complesso farmaco-proteina plasmatici, portando però a due effetti collaterali: sia all’eliminazione di un farmaco attivo, sia all’abbassamento della concentrazione di proteine plasmatiche.

37
Legame alle proteine Soprattutto alle albumine
Il farmaco legato non attraversa le membrane Equilibrio continuo tra parte libera e legata Farmaci molto legati... Legati alle albumine o alle glicoproteine alfa: FANS warfarin ceftiofur doxiciclina furosemide chinidina diazepam propranololo …

38
Fattori che modificano il legame farmaco-proteico
Ogni modificazione del tasso di proteine plasmatiche: Insufficienza epatica Insufficienza renale Enteropatie Parassitosi Ustioni Se aumenta la quota libera: Aumento dell’effetto Aumento della velocità di eliminazione

39
BARRIERE CELLULARI Tutti i tessuti sono estesamente irrorati dai capillari sanguigni. Vi è quindi un’ampia superficie di scambio tra sangue e tessuti. In genere, tra le cellule dell’endotelio capillare sono presenti dei pori, che permettono il passaggio dal sangue al tessuto anche di molecole non lipofile. I pori hanno un diametro di circa 10 nm, che limita o impedisce il passaggio di molte proteine. In alcuni organi (SNC, occhio, placenta), l’endotelio capillare è praticamente privo di pori. Inoltre, vi è scarsissima attività di endocitosi. Nel cervello, inoltre, i capillari sono rivestiti da cellule gliali. A questo livello vi è quindi una barriera al passaggio di sostanze che non diffondono attraverso la membrana cellulare. Farmaci non lipofili non penetrano significativamente nel SNC e non hanno quindi effetti centrali (farmaco-terapeutici). Es. antimuscarinici di sintesi.
, l’endotelio capillare è praticamente privo di pori. Inoltre, vi è scarsissima attività di endocitosi. Nel cervello, inoltre, i capillari sono rivestiti da cellule gliali. A questo livello vi è quindi una barriera al passaggio di sostanze che non diffondono attraverso la membrana cellulare. Farmaci non lipofili non penetrano significativamente nel SNC e non hanno quindi effetti centrali (farmaco-terapeutici). Es. antimuscarinici di sintesi.")
40
Barriera ematoencefalica
Non rappresenta un ostacolo assoluto al passaggio degli xenobiotici nel sistema nervoso centrale, ma fattori anatomici e fisiologici ne riducono la permeabilità; Il liquido interstiziale nel SNC ha una composizione nettamente diversa da quella di altri tessuti; in particolare sono praticamente assenti le proteine plasmatiche. A livello della barriera emato-encefalica (e di altre barriere) vi sono numerosi sistemi di trasporto, che consentono il passaggio di sostanze non lipofile necessarie per il metabolismo del SNC (es. glucosio, aminoacidi). le cellule endoteliali dei capillari cerebrali hanno giunzioni serrate e i pori sono virtualmente assenti le cellule endoteliali stesse contengono un carrier proteico ATP-dipendente in grado di trasportare alcune sostanze in direzione del sangue i capillari del sistema nervoso centrale sono in gran parte avvolti dai processi delle cellule gliali-
 vi sono numerosi sistemi di trasporto, che consentono il passaggio di sostanze non lipofile necessarie per il metabolismo del SNC (es. glucosio, aminoacidi). le cellule endoteliali dei capillari cerebrali hanno giunzioni serrate e i pori. sono virtualmente assenti. le cellule endoteliali stesse contengono un carrier proteico ATP-dipendente. in grado di trasportare alcune sostanze in direzione del sangue. i capillari del sistema nervoso centrale sono in gran parte avvolti dai processi. delle cellule gliali-")
41
Nell’encefalo circola il liquido cefalo-rachidiano (LCR, liquor), che origina dal sangue a livello dei plessi coroidei, dove l’endotelio capillare è più fenestrato ed è molto più permeabile che in altre regioni del SNC. Farmaci idrofili possono quindi passare nel LCR. Tuttavia, data la bassa estensione del circolo del LCR, il passaggio nel liquido interstiziale è limitato. Esistono inoltre sistemi di trasporto LCR sangue. Le zone del SNC vicine ai plessi coroidei e alcune regioni periventricolari sono meno protette dalla barriera emato-encefalica e più sensibili ai farmaci presenti nel sangue (es. CTZ, Chemoreceptor Trigger Zone, l’area di ‘innesco’ del riflesso del vomito). In condizioni patologiche (infezioni con infiammazione delle meningi, febbre elevata, aterosclerosi) si ha una compromissione anatomico-funzionale della barriera emato-encefalica, con aumento della permeabilità.
. In condizioni patologiche (infezioni con infiammazione delle meningi, febbre elevata, aterosclerosi) si ha una compromissione anatomico-funzionale della barriera emato-encefalica, con aumento della permeabilità.")
42
La barriera placentare
Barriera emato-testicolare Localizzata tra il lume del capillare interstiziale e il lume del tubulo seminifero è costituita da endotelio capillare, lamina basale capillare, endotelio linfatico, cellule mioidi, lamina basale del tubulo seminifero e cellule del Sertoli. La barriera placentare Il sangue fetale è separato da quello materno dal sincizio placentare, dall’interstizio villare e dalle cellule endoteliali dei capillari villari. A livello del sincizio placentare sono presenti molti sistemi di trasporto e di endocitosi mediata da recettore (es. transferrina, IgG, peptidi, proteine) La barriera placentare è meno selettiva di quella emato-encefalica. Inoltre, il sangue materno circola molto lentamente aumenta il tempo di scambio. Quasi tutti i farmaci (tranne farmaci idrofili di grosse dimensioni molecolari) possono raggiungere il feto, anche se lentamente e a concentrazioni più basse rispetto a quelle materne.
 La barriera placentare è meno selettiva di quella emato-encefalica. Inoltre, il sangue materno circola molto lentamente aumenta il tempo di scambio. Quasi tutti i farmaci (tranne farmaci idrofili di grosse dimensioni molecolari) possono raggiungere il feto, anche se lentamente e a concentrazioni più basse rispetto a quelle materne.")
43
Barriera placentare Protegge il feto da sostanze nocive presenti nel sangue materno, ma deve garantire il passaggio di numerose sostanze; processi di trasporto attivo consentono il passaggio di sostanze nutritive e vitamine dalla madre al feto. Consiste di numerosi strati di cellule interposti tra la circolazione fetale e quella materna, strati che variano con il periodo di gestazione e da una specie all’altra TESSUTI MATERNI TESSUTI FETALI endotelio tess.conn. epitelio trofoblasto tess.conn. endotelio Specie Epiteliocoriale Sindesmocoriale Endoteliocoriale Emocoriale Emoendocoriale Maiale, cavallo Bovini, ovini Cane, gatto Uomo, scimmia Ratto, coniglio

44
I meccanismi di eliminazione di un farmaco sono:
-la sua biotrasformazione (metabolismo) -la sua escrezione Anche se uno dei due predomina sull’altro, sia il metabolismo epatico che l’escrezione renale cooperano allontanando il farmaco dall’organismo. Il destino di un farmaco dipende da alcune sue caratteristiche quali la liposolubilità ed il grado di ionizzazione. Il requisito liposolubilità è primario per la biotrasformazione da parte del sistema enzimatico microsomiale, mentre i farmaci polari e la maggior parte dei metaboliti vengono escreti dal rene. Oltre che nel fegato un metabolismo dei farmaci può realizzarsi nel plasma ematico e nel lume dei visceri (reazioni di idrolisi e di riduzione), ma anche in altri tessuti (rene, polmone).
 -la sua escrezione. Anche se uno dei due predomina sull’altro, sia il metabolismo epatico che l’escrezione renale cooperano allontanando il farmaco dall’organismo. Il destino di un farmaco dipende da alcune sue caratteristiche quali la liposolubilità ed il grado di ionizzazione. Il requisito liposolubilità è primario per la biotrasformazione da parte del sistema enzimatico microsomiale, mentre i farmaci polari e la maggior parte dei metaboliti vengono escreti dal rene. Oltre che nel fegato un metabolismo dei farmaci può realizzarsi nel plasma ematico e nel lume dei visceri (reazioni di idrolisi e di riduzione), ma anche in altri tessuti (rene, polmone).")
45
(fase II)-->PRODOTTI CONIUGATI
BIOTRASFORMAZIONI Nell’organismo i farmaci subiscono trasformazioni metaboliche volte alla formazione di metaboliti con proprietà chimico-fisiche favorevoli ad una successiva escrezione; questi prodotti, di solito, sono meno liposolubili e molto polari, ciò li rende suscettibili di processi escretivi dipendenti da appositi trasportatori. FARMACO-->REAZIONI (fase I) OSSIDATIVE-RIDUTTIVE-IDROLITICHE---> METABOLITA ---> REAZIONI DI SINTESI (fase II)-->PRODOTTI CONIUGATI Le trasformazioni di prima fase smascherano o introducono nella molecola del farmaco raggruppamenti polari (OH-COOH-NH2 ) che rendono il composto idoneo a subire processi di coniugazione con sostanze endogene ( ac.glucuronico, acetato, solfato ed aminoacidi diversi) con formazione di composti idrosolubili e sempre privi di attività farmacologica.
 OSSIDATIVE-RIDUTTIVE-IDROLITICHE---> METABOLITA ---> REAZIONI DI SINTESI. (fase II)-->PRODOTTI CONIUGATI. Le trasformazioni di prima fase smascherano o introducono nella molecola del farmaco raggruppamenti polari (OH-COOH-NH2 ) che rendono il composto idoneo a subire processi di coniugazione con sostanze endogene ( ac.glucuronico, acetato, solfato ed aminoacidi diversi) con formazione di composti idrosolubili e sempre privi di attività farmacologica.")
46
Il metabolismo di uno xenobiotico è l’insieme delle trasformazioni chimiche che la molecola subisce all’interno dell’organismo, principalmente ad opera di enzimi. Il metabolismo ha una ‘logica’: trasformare la molecola in modo che possa essere facilmente escreta.

47
Metabolismo ed attività biologica
I metaboliti sono molecole con caratteristiche chimiche e tossicologiche diverse dal composto di partenza. Se lo xenobiotico agisce interagendo con recettori (es., atropina, cianuro, organofosforici), i metaboliti sono in genere privi di attività biologica. In questo caso, il metabolismo è quindi una modalità di ‘eliminazione’. Diversi xenobiotici (ad es. molti farmaci), tuttavia, hanno metaboliti ‘attivi’, dotati cioè di attività farmacologica simile a quella del composto ‘madre’.
, i metaboliti sono in genere privi di attività biologica. In questo caso, il metabolismo è quindi una modalità di ‘eliminazione’. Diversi xenobiotici (ad es. molti farmaci), tuttavia, hanno metaboliti ‘attivi’, dotati cioè di attività farmacologica simile a quella del composto ‘madre’.")
48
Molti xenobiotici (dotati o meno di attività propria) sono trasformati in composti potenzialmente tossici dal metabolismo bioattivazione; es.: benzene, IPA, idrocarburi alogenati, aflatossine. I metaboliti tossici possono essere ulteriormente metabolizzati, con formazione di composti non tossici (detossificazione). Il metabolismo degli xenobiotici è estremamente importante in Tossicologia, poiché può avere sia un effetto protettivo (eliminazione e/o detossificazione dello xenobiotico) sia un effetto dannoso (formazione di metaboliti tossici).
. Il metabolismo degli xenobiotici è estremamente importante in Tossicologia, poiché può avere sia un effetto protettivo (eliminazione e/o detossificazione dello xenobiotico) sia un effetto dannoso (formazione di metaboliti tossici).")
49
La presenza di particolari gruppi funzionali nella molecola di un farmaco lascia prevedere, in linea di massima, la via metabolica che esso seguirà nell’organismo. Anche se le reazioni di prima fase generano di solito composti meno attivi, in alcuni casi può accadere che il metabolica formatosi conservi l’attività piena della molecola originaria o addirittura che acquisisca attività maggiori rispetto a quella.

50
PRINCIPALI VIE DI BIOTRASFORMAZIONE A = citocromo P450 ossidato
Reazioni di I fase: la reazione più importante nel metabolismo dei farmaci liposolubili e di ormoni steroidei è quella di ossidazione da parte di enzimi microsomiali, particolarmente esigenti di NADPH e di ossigeno molecolare (detti ossidasi a funzione mista). La capacità di catalizzare una grande quantità di reazioni ossidative può essere attribuito ad un comune meccanismo: l’idrossilazione che comporta la riduzione di un costituente microsomiale, il citocromo P450 , un vero e proprio enzima ossidante. NADPH + A + H+ AH2 +NADP+ AH2 + O2 COMPLESSO OSSIDATO ATTIVO COMPL.OSSID. ATTIVO + FARMACO FARMACO OSSIDATO +A + H2O A = citocromo P ossidato AH = citocromo P ridotto
. La capacità di catalizzare una grande quantità di reazioni ossidative può essere attribuito ad un comune meccanismo: l’idrossilazione che comporta la riduzione di un costituente microsomiale, il citocromo P450 , un vero e proprio enzima ossidante. NADPH + A + H+ AH2 +NADP+ AH2 + O2 COMPLESSO OSSIDATO ATTIVO. COMPL.OSSID. ATTIVO + FARMACO FARMACO OSSIDATO +A + H2O. A = citocromo P450 ossidato. AH = citocromo P450 ridotto.")
51
Le reazioni ossidative più importanti comprendono:
idrossilazione di composti aromatici ossidazione di composti alifatici dealchilazione di radicali agganciati all’ O oppure all’ N deaminazioni ossidative desulfurazioni ( sostituzione di un atomo di S con uno di O ) ossidazione dell’atomo di S Non è raro il caso in cui il farmaco sia metabolizzato con due o più procedure ed in tal caso, le quote di metaboliti formatisi dipendono dalle attività differenziali di vari sistemi enzimatici metabolizzanti (anfetamina, fenilbutazone).
 ossidazione dell’atomo di S. Non è raro il caso in cui il farmaco sia metabolizzato con due o più procedure ed in tal caso, le quote di metaboliti formatisi dipendono dalle attività differenziali di vari sistemi enzimatici metabolizzanti (anfetamina, fenilbutazone).")
52
Le reazioni metaboliche non microsomiali più importanti sono :
I microsomi epatici sono anche in grado di ridurre i composti azotati e quelli nitrati alle corrispondenti amine, necessitando di ambiente anaerobico, di NADPH e di enzimi di natura flavoproteica. Anche la dealogenazione riduttiva di alcuni composti anestetici generali (alotano, metossiflurano) è quasi certamente dovuta ai sistemi enzimatici microsomiali. Le reazioni metaboliche non microsomiali più importanti sono : ossidazione degli alcoli e delle aldeidi riduzione di chetoni deaminazione sostenuta da monoaminoossidasi (MAO) idrolisi degli esteri e delle amidi (più veloce per gli esteri e meno per le amidi). I microrganismi del rumine ed i batteri intestinali sostengono reazioni di idrolisi e di riduzione.
 è quasi certamente dovuta ai sistemi enzimatici microsomiali. Le reazioni metaboliche non microsomiali più importanti sono : ossidazione degli alcoli e delle aldeidi. riduzione di chetoni. deaminazione sostenuta da monoaminoossidasi (MAO) idrolisi degli esteri e delle amidi (più veloce per gli esteri e meno per le amidi). I microrganismi del rumine ed i batteri intestinali sostengono reazioni di idrolisi e di riduzione.")
53
Reazioni di II fase (sintesi): possono avvenire quando un farmaco o il suo metabolica di prima fase contengono gruppi funzionali del tipo ossidrile (-OH), carbossile (-COOH), aminico (-NH2) o sulfidrilico (-SH) disponibili a combinarsi con un composto naturale prodotto dall’organismo. Possono così formarsi nuovi metaboliti idrosolubili e polari prontamente escretabili. Le sostanze coniuganti principali sono: ac.glucuronico, glicina, cisteina, metionina (metilazione), il radicale solfato (esteri solfati), il radicale acetato (acetilazione). Tali coniuganti non reagiscono direttamente col farmaco o col suo metabolita di prima fase ma con forme previamente inattivate o attive di esso. Le forme attivate sono dovute alla presenza di nucleotidi e la reazione tra nucleotide, farmaco e coniugante viene catalizzata da enzimi. Diversamente dalle reazioni di prima fase, alcune reazioni di coniugazione appaiono limitate o assenti in alcune specie animali.
, il radicale solfato (esteri solfati), il radicale acetato (acetilazione). Tali coniuganti non reagiscono direttamente col farmaco o col suo metabolita di prima fase ma con forme previamente inattivate o attive di esso. Le forme attivate sono dovute alla presenza di nucleotidi e la reazione tra nucleotide, farmaco e coniugante viene catalizzata da enzimi. Diversamente dalle reazioni di prima fase, alcune reazioni di coniugazione appaiono limitate o assenti in alcune specie animali.")
54
La formazione di glucuronidi è un processo metabolico fondamentale per certi farmaci e per taluni composti endogeni (ormoni steroidei, tiroxina, bilirubina) e porta alla formazione di composti più idrosolubili (più intensamente ionizzati ai valori di pH fisiologici); tutto ciò ne facilita l’allontanamento dall’organismo, riducendone la distribuzione extravascolare, rendendoli più idonei ad una secrezione renale e biliare assicurata da trasportatori. I glucuronidi escreti con la bile possono subire idrolisi in sede intestinale mediante l’azione delle β-glucuronidasi, liberando il farmaco che può essere riassorbito e dar luogo ad un circolo entero-epatico. Non tutti i glucuronidi sono idrolizzati dalle β-glucuronidasi enteriche, esistono anche differenze specie–specifiche e tappe coniugative alternative, come per gli insetti dove è stata descritta una coniugazione con β-glicosidi.

55
BIOTRASFORMAZIONI SOSTENUTE DA MICRORGANISMI
La solfoconiugazione è una via metabolica importante, alternativa alla glucuronidazione, per la biotrasformazione di fenoli e alcoli alifatici (fenolo, acetaminofene, morfina, isoprenalina, ac.ascorbico), ma anche di sostanze endogene (condroitina, eparina, alcuni steroidi). L’acetilazione di tutti i tipi di amino-gruppi è una reazione possibile in molte specie animali (eccetto cane e volpe); è la via più importante di biotrasformazione dei sulfamidici. BIOTRASFORMAZIONI SOSTENUTE DA MICRORGANISMI La microflora presente nell’apparato digerente è in grado di sostenere numerose trasformazioni metaboliche, soprattutto idrolitiche e reduttasiche. Il metabolismo microbico può avvenire dopo somministrazione orale di un farmaco o dopo diffusione passiva dello stesso allo stato non ionizzato dal circolo sistemico. I farmaci coniugati, riversati con la bile nel tubo digerente, vengono idrolizzati da β-glucuronidasi batteriche ( soprattutto prodotte da E.Coli ) e di poi riassorbite instaurando il circolo entero-epatico.
, ma anche di sostanze endogene (condroitina, eparina, alcuni steroidi). L’acetilazione di tutti i tipi di amino-gruppi è una reazione possibile in molte specie animali (eccetto cane e volpe); è la via più importante di biotrasformazione dei sulfamidici. BIOTRASFORMAZIONI SOSTENUTE DA MICRORGANISMI. La microflora presente nell’apparato digerente è in grado di sostenere numerose trasformazioni metaboliche, soprattutto idrolitiche e reduttasiche. Il metabolismo microbico può avvenire dopo somministrazione orale di un farmaco o dopo diffusione passiva dello stesso allo stato non ionizzato dal circolo sistemico. I farmaci coniugati, riversati con la bile nel tubo digerente, vengono idrolizzati da β-glucuronidasi batteriche ( soprattutto prodotte da E.Coli ) e di poi riassorbite instaurando il circolo entero-epatico.")
56
Poiché i microrganismi gastrointestinali possono trasformare alcuni farmaci, la somministrazione prolungata di antibatterici può alterare tale funzione, indipendentemente dalla via di somministrazione dell’antibatterico, portando ad una depressione della flora batterica saprofita tanto maggiore a seconda del grado di assorbibilità dell’antibatterico da parte del piccolo intestino o, nel caso di somministrazione parenterale, dalla quota di dose diffusa nel grosso intestino e nel rumine; tale funzione può essere condizionata anche dal grado di liposolubilità e di ionizzazione del farmaco stesso. MODIFICAZIONI DELLE CAPACITA’ METABOLICHE PRODOTTE DAI FARMACI Il metabolismo di un farmaco ne facilita in linea di massima l’eliminazione, per cui alterazioni del metabolismo potranno influenzare la durata d’azione del farmaco stesso. I fattori che possono modificare le capacità metaboliche dell’organismo sono: alcuni farmaci liposolubili alcune sostanze chimiche diffuse nell’ambiente ( pesticidi e carcinogeni) riduzioni del flusso di sangue distrettuale al fegato.
 riduzioni del flusso di sangue distrettuale al fegato.")
57
Alcuni farmaci, tuttavia, sono in grado di stimolare i sistemi microsomiali epatici provocando un aumento del patrimonio enzimatico piuttosto che un potenziamento di attività specifica e questo fenomeno prende il nome di induzione enzimatica mentre i farmaci che la provocano sono detti induttori. Ogni farmaco liposolubile a PH fisiologico è potenzialmente in grado di provocare induzione se somministrato continuativamente. L’influenza del fenomeno induttivo sull’azione farmacologica di un farmaco dipende dall’attività relativa del farmaco stesso e dei suoi prodotti di reazione (metaboliti di I fase).
.")
58
insetticidi organofosforati tetracloruro di carbonio
Il metabolismo di un farmaco può anche essere ridotto per inibizione del processo biotrasformativo, assistendo in questo caso ad una ritardata eliminazione del farmaco. Se si instaura l’inibizione di una via metabolica, può prevalere una seconda e ciò può essere molto importante soprattutto se il farmaco è dotato di potenziale di tossicità. Tra i farmaci che possono portare ad inibizione dell’attività microsomiale epatica sono da ricordare : cimetidina cloramfenicolo chinidina insetticidi organofosforati tetracloruro di carbonio in ragione di ciò bisogna porre attenzione ogni qual volta si usano più farmaci per un’unica terapia.

59
ESCREZIONE DEI FARMACI
La gran parte dei farmaci viene eliminata per azione combinata dei processi di biotrasformazione e di escrezione . La biotrasformazione riduce la liposolubilità dei farmaci, per cui i loro metaboliti vengono escreti rapidamente. Sebbene il rene sia l’organo escretore più importante, taluni composti vengono escreti anche con la bile. Vie di escrezione alternative a quella renale sono quella epatica, salivare, sudorifera, mammaria e polmonare. L’escrezione polmonare comporta la diffusione di sostanze volatili, dal circolo sistemico negli spazi alveolari polmonari, dai quali successivamente esse vengono allontanate tramite l’espirazione.

60
Escrezione renale E’ il principale meccanismo di eliminazione dei farmaci, prevalentemente ionizzati a valori di pH fisiologici e di quelli dotati di bassa liposolubilità. Fra i farmaci che sono eliminati con le urine senza alcuna trasformazione sono da ricordare: molti antibiotici (penicilline, con l’eccezione della nafcillina; cefalosporine; aminoglicosidici; ossitetraciclina) la maggior parte dei diuretici (escluso l’acido etacrinico) agenti di blocco neuromuscolare con meccanismo competitivo ( d-tubocurarina, gallamina) e, probabilmente, il glucoside cardioattivo digossina.
 la maggior parte dei diuretici (escluso l’acido etacrinico) agenti di blocco neuromuscolare con meccanismo competitivo. ( d-tubocurarina, gallamina) e, probabilmente, il glucoside cardioattivo digossina.")
61
Il Nefrone Struttura dei segmenti tubulari
Tubulo contorto prossimale Ansa discendente o di Henle (segmento sottile) Ansa ascendente o di Henle (segmento sottile e spesso) Capsula glomerulare o di Bowman Dotto collettore midollare Dotto collettore corticale Tubulo contorto distale
 Ansa ascendente o di Henle (segmento sottile e spesso) Capsula glomerulare o di Bowman. Dotto collettore midollare. Dotto collettore corticale. Tubulo contorto distale.")
62
filtrazione glomerulare, per le molecole libere nel plasma
In sede renale, i farmaci ed i loro metaboliti, vengono sottoposti, a seconda delle proprietà chimico-fisiche delle varie sostanze, a: filtrazione glomerulare, per le molecole libere nel plasma escrezione, per i composti organici polari, dipendente da trasportatori, da parte delle cellule del tubulo contorto prossimale riassorbimento passivo pH dipendente, per diffusione non ionica, per le sostanze liposolubili nelle porzione distale del nefrone ( soprattutto elettroliti organici deboli). Non è possibile prevedere quanto il legame proteico contribuisca ad ostacolare la filtrazione glomerulare di un farmaco; comunque, una percentuale di legame superiore all’80% ne impedisce certamente il passaggio attraverso la membrana porosa del capillare glomerulare. La quantità di farmaco che perviene nell’ultrafiltrato è funzione della sua concentrazione libera nel plasma e dell’entità di filtrazione glomerulare ( GFR ).
. Non è possibile prevedere quanto il legame proteico contribuisca ad ostacolare la filtrazione glomerulare di un farmaco; comunque, una percentuale di legame superiore all’80% ne impedisce certamente il passaggio attraverso la membrana porosa del capillare glomerulare. La quantità di farmaco che perviene nell’ultrafiltrato è funzione della sua concentrazione libera nel plasma e dell’entità di filtrazione glomerulare. ( GFR ).")
63
In ogni caso, l’elevata percentuale di legame alle proteine plasmatiche non impedisce l’escrezione tubulare dei farmaci perchè il complesso farmaco-albumina si dissocia man mano che avviene l’allontanamento della quota libera di farmaco dal plasma. La contemporanea somministrazione di due farmaci (acidi o basici), che utilizzino una modalità di escrezione mediante gli stessi trasportatori, ritarda l’escrezione della sostanza, meno velocemente trasportata, come accade nel caso del probenecid che rallenta l’eliminazione della penicillina G, limitandone l’escrezione tubulare, e ciò a riprova che un certo tipo di trasporto si avvale di trasportatori.
, che utilizzino una modalità di escrezione mediante gli stessi trasportatori, ritarda l’escrezione della sostanza, meno velocemente trasportata, come accade nel caso del probenecid che rallenta l’eliminazione della penicillina G, limitandone l’escrezione tubulare, e ciò a riprova che un certo tipo di trasporto si avvale di trasportatori.")
64
Anche se un farmaco può pervenire nel liquido tubulare per filtrazione glomerulare o per escrezione delle cellule del tubulo contorto prossimale, l’entità di una sua purificazione renale risulta esigua; ciò può essere motivato da un significativo riassorbimento da parte del distretto tubulare del nefrone. Siccome il riassorbimento tubulare avviene per diffusione passiva, solo la quota liposolubile non ionizzata dell’elettrolita organico debole può essere riassorbita. Il grado di riassorbimento è condizionato dalla concentrazione del farmaco e del suo stato di ionizzazione nel liquido che transita nel tubulo distale.

65
Ciò avviene quando si procede all’alcalinizzazione delle urine o quando si stimola una diuresi alcalina per facilitare l’escrezione di composti organici acidi eventualmente somministrati in eccesso, a condizione che una cospicua quota della quantità di farmaco presente nell’organismo sia escreta con le urine in forma immodificata. Il pH normale delle urine dei carnivori è acido, quello degli erbivori è alcalino, ma risulta condizionato dal regime alimentare. Gli erbivori poppanti eliminano in genere urina acida, almeno finchè sono alimentati solo con latte

66
I farmaci liposolubili tendono ad essere escreti a concentrazioni
ELIMINAZIONE PER VIA RENALE I farmaci liposolubili tendono ad essere escreti a concentrazioni simili a quelle presenti nel plasma. La loro concentrazione dipende soprattutto dal volume delle urine 2) I farmaci polari tendono ad essere escreti nelle urine a concentrazioni superiori a quelle presenti nel plasma , quindi la loro escrezione dipende più dal volume del filtrato glomerulare che dal volume delle urine 3) I farmaci coniugati si comportano in maniera simile alle sostanze polari, ma possono essere escreti in misura maggiore perché soggetti a meccanismi di secrezione attiva 4) I farmaci che si ionizzano facilmente, cioè acidi e basi, vengono escreti in maniera pH dipendente
 I farmaci polari tendono ad essere escreti nelle urine a concentrazioni. superiori a quelle presenti nel plasma , quindi la loro escrezione. dipende più dal volume del filtrato glomerulare che dal volume. delle urine. 3) I farmaci coniugati si comportano in maniera simile alle sostanze. polari, ma possono essere escreti in misura maggiore perché. soggetti a meccanismi di secrezione attiva. 4) I farmaci che si ionizzano facilmente, cioè acidi e basi, vengono. escreti in maniera pH dipendente.")
67
presenza nella molecola di gruppi polari
ESCREZIONE BILIARE Le peculiarità che deve avere un composto per essere suscettibile di escrezione biliare sono: PM > 300 Da presenza nella molecola di gruppi polari La coniugazione con acido glucuronico può essere un fattore condizionante l’escrezione biliare di un farmaco, di metaboliti di prima fase o di talune sostanze endogene. L’escrezione biliare è un importante meccanismo di eliminazione per anioni e cationi organici che non siano riassorbibili in sede intestinale. Alcuni farmaci (nafcillina, eritromicina, digitossina), l’acido iopanoico, alcune sostanze endogene di tipo steroideo e i glicuronidi di molti composti (CAF, morfina, bilirubina) vengono escreti in misura elevata con la bile.
, l’acido iopanoico, alcune sostanze endogene di tipo steroideo e i glicuronidi di molti composti (CAF, morfina, bilirubina) vengono escreti in misura elevata con la bile.")
68
escretrici biliari efficienti: ratto, cane e pollo
L’importanza di questa via dipende dal tipo di sostanza e dalle capacità escretive delle singole specie animali che, all’uopo, vengono classificate in : escretrici biliari efficienti: ratto, cane e pollo escretrici biliari moderatamente efficienti: gatto e ovino escretrici biliari scarsamente efficienti: cavia, coniglio e scimmia Questa categorizzazione è rivolta a sostanze con PM minimo, mentre la variabilità di specie è notevole al crescere del PM. Quando quest’ultimo arriva a 500, come può accadere ad un glicuronide, esso sarà escreto con la bile da parte di tutte le specie animali.

69
Escrezione biliare Acidi Basi Sostanze neutre Metalli 4 sistemi di trasporto attivo P.M. < 250 eliminazione renale P.M. > 500 eliminazione biliare Composti polari 250 < P.M. < 500 BUONI MODESTI POVERI cane, ratto, uccelli gatto, pecora cavia, coniglio, scimmia

70
Il rapporto tra la misura contemporanea di farmaco totale nel plasma e
Escrezione salivare Bassa percentuale di proteine Farmaco prevalentemente non legato Il rapporto tra la misura contemporanea di farmaco totale nel plasma e nella saliva fornisce una buona indicazione della percentuale di farmaco libero nel plasma pH saliva 8 –8.4 ruminanti cavallo, cane, gatto uomo

71
I composti escreti con la bile pervengono al piccolo intestino, dove, a seconda della loro liposolubilità, alcuni vengono riassorbiti, altri vengono idrolizzati dalle β-glicuronidasi e possono, anch’essi, essere riassorbiti (circolo entero-epatico). Quando una quota significativa della dose somministrata è soggetta al circolo entero - epatico, si avrà un rallentamento dell’eliminazione del farmaco, specie se essa avviene tramite escrezione renale.

72
CONCENTRAZIONI PLASMATICHE TERAPEUTICHE
I principali fattori che condizionano il livello di concentrazione plasmatica di un farmaco sono: l’entità della dose il tipo di formulazione della preparazione del farmaco la via di somministrazione la disponibilità sistemica del farmaco e il suo grado di assorbibilità la capacità di distribuzione e di legame alle proteine plasmatiche la velocità della sua eliminazione

73
Inoltre, è da considerare la capacità del farmaco a raggiungere i siti dove agire, il che comporta una permeazione dei tessuti, ed è elemento condizionante la concentrazione di farmaco che si ottiene nella biofase; il tutto serve a determinare l’intensità della risposta osservabile. La gamma di concentrazioni terapeutiche, per un farmaco antibatterico, indica le concentrazioni plasmatiche che dovrebbero risultare efficaci nei confronti dei microrganismi più sensibili e nello stesso tempo atossiche per gli animali. Questa gamma tiene conto della MIC per l’agente patogeno e del margine di sicurezza caratteristico del farmaco.

74
SOMMINISTRAZIONE DEI FARMACI
Un trattamento terapeutico può comportare la somministrazione di una sola dose di farmaco o di dosi multiple a tempi distanziati fissi e deve indicare la dose per unità di tempo. Quando la frequenza di somministrazione è impraticabile o passibile di generare effetti tossici, il farmaco dovrebbe essere somministrato in preparazioni a lento rilascio o per infusione IV continua, il che consente di apportare aggiustamenti della dose in funzione della risposta osservata. In terapia antibatterica, il miglior indice di efficacia del trattamento è la guarigione dall’infezione batterica. Una risposta soddisfacente rassicura che il farmaco antibatterico è stato somministrato a dosaggio idoneo e per un tempo sufficiente.

75
IL PASSAGGIO DEI FARMACI NEL LATTE
Pone problemi rilevanti nella terapia veterinaria, essendo il latte uno degli alimenti più importanti per la specie umana sia come tale che sotto forma di prodotti di trasformazione. I fattori condizionanti il passaggio dei farmaci nel latte sono: -costituzione chimica del farmaco stesso -dose somministrata e durata del trattamento -via di somministrazione (se per curare mastiti si somministra il farmaco per via endomammaria avrà persistenza maggiore rispetto ad una somministrazione generale)
")
76
La presenza di farmaci nel latte è determinata dai trattamenti terapeutici :
-endomammari in animali in lattazione -endomammari in animali in asciutta -parenterali in animali in lattazione La somministrazione di farmaci a fini auxinici non sembra creare problemi, sia per la salute umana sia alle implicazioni industriali (caseificazione del latte e maturazione dei formaggi). E’ da notare inoltre che, a fini auxinici, i chemioantibiotici si somministrano, generalmente ad animali che ancora non producono latte.
. E’ da notare inoltre che, a fini auxinici, i chemioantibiotici si somministrano, generalmente ad animali che ancora non producono latte.")
77
ANTIBIOTICI NEL LATTE L’antibiotico più frequentemente rinvenuto nel latte di bovino è stato la penicillina G. E’ stato osservato che è piuttosto raro reperire antibiotici oltre la 132a h dall’ultimo trattamento endomammario, però, diversi fattori possono variare la permanenza nella mammella e quindi nel latte; tra essi importanti sono: il veicolo (maggiore la presenza di antibiotico se il veicolo è oleoso piuttosto che acquoso, poiché il latte presenta una certa quantità di grassi) e la quantità di latte prodotto (una produzione < 9 l comporta un’ulteriore rallentata escrezione).
 e la quantità di latte prodotto (una produzione. < 9 l comporta un’ulteriore rallentata escrezione).")
78
E’ da notare che, anche se la somministrazione del farmaco viene effettuata in un quarto, l’attività antibiotica può essere riscontrata nei quarti non trattati, così come nel sangue. Ciò perché il farmaco somministrato in un quarto, viene assorbito, passa in circolo, viene escreto per le comuni vie di eliminazione ma viene secreto negli altri quarti (tranne che per la streptomicina). Il latte ottenuto dai quarti non trattati può essere privo di attività antibiotica prima di quello proveniente dal quarto trattato. Attraverso il circolo sanguigno, i farmaci arrivano nell’interstizio perialveolare della ghiandola mammaria. Da qui, durante la lattazione, le molecole libere, non più legate alle proteine plasmatiche (quindi attivate), possono attraversare la lamina basale e l’epitelio alveolare per giungere nel latte, per mezzo di diversi meccanismi
, possono attraversare la lamina basale e l’epitelio alveolare per giungere nel latte, per mezzo di diversi meccanismi.")
79
Nei processi di diffusione passiva, la membrana alveolare può essere paragonata ad un cribro con larghi fori acquosi attraverso i quali passano liberamente le molecole disciolte nel liquido; in alcuni casi tale diffusione è facilitata da “carriers” ma comunque diffondono meglio le sostanze più idrosolubili. Nei processi di attraversamento per adsorbimento e solubilizzazione nelle fasi lipoidee, la membrana alveolare si comporta come se fosse costituita da uno strato lipidico nel quale i farmaci liposolubili si solubilizzano per diffondere poi dall’altro lato. L’equilibrio si raggiunge all’ottenimento di eguali concentrazioni di farmaco liposolubile non ionizzato ai due lati della membrana.

80
Trasporto con proteine carriers
Altri meccanismi che permettono ai farmaci di passare dal sangue al latte sono quelli attivi, che richiedono dispendi di energia (consumo di ATP), fornita da processi metabolici intracellulari; i farmaci attraversano le membrane o legati a proteine carriers o per pinocitosi. Trasporto con proteine carriers E’ un processo simile a quello della diffusione facilitata ma consente di trasferire il farmaco anche verso il lato con concentrazione più elevata ed è selettivo e saturabile. Pinocitosi Nel processo di pinocitosi, il farmaco è inglobato in un'invaginazione della membrana cellulare e trasportato all’altro lato della stessa; quindi la parte invaginata si distacca dalla superficie cellulare con il farmaco che rimane inglobato nella membrana.
, fornita da processi metabolici intracellulari; i farmaci attraversano le membrane o legati a proteine carriers o per pinocitosi. Trasporto con proteine carriers. E’ un processo simile a quello della diffusione facilitata ma consente di trasferire il farmaco anche verso il lato con concentrazione più elevata ed è selettivo e saturabile. Pinocitosi. Nel processo di pinocitosi, il farmaco è inglobato in un invaginazione della membrana cellulare e trasportato all’altro lato della stessa; quindi la parte invaginata si distacca dalla superficie cellulare con il farmaco che rimane inglobato nella membrana.")
81
RIPERCUSSIONI SULLA SALUTE PER LA PRESENZA DEI FARMACI NEL LATTE
I chemio-antibiotici presenti nel latte possono provocare le medesime manifestazioni tossiche alle quali danno luogo durante le terapie generali, soprattutto nei neonati umani e nei cuccioli animali che vengono alimentati con latte proveniente da soggetti trattati. Le manifestazioni più importanti e più numerose sono quelle allergiche (dermatiti, shock anafilattico) e l’antibiotico funge da agente sensibilizzante o da agente scatenante. Altro inconveniente, specie nei lattanti, è la possibilità di determinare resistenza batterica o, nei neonati, soprattutto nei prematuri, la possibilità di spostare la bilirubina legata alle proteine e provocare una sintomatologia piuttosto grave (la sindrome grigia). Pertanto, anche se i chemio-antibiotici determinano nel latte concentrazioni molto basse, è sempre opportuno evitare di utilizzare questi farmaci negli animali che allattano o che producono latte per uso umano.
 e l’antibiotico funge da agente sensibilizzante o da agente scatenante. Altro inconveniente, specie nei lattanti, è la possibilità di determinare resistenza batterica o, nei neonati, soprattutto nei prematuri, la possibilità di spostare la bilirubina legata alle proteine e provocare una sintomatologia piuttosto grave (la sindrome grigia). Pertanto, anche se i chemio-antibiotici determinano nel latte concentrazioni molto basse, è sempre opportuno evitare di utilizzare questi farmaci negli animali che allattano o che producono latte per uso umano.")
82
INCONVENIENTI NEI PRODOTTI DI TRASFORMAZIONE DEL LATTE
La presenza di chemio-antibiotici a determinate concentrazioni modifica profondamente l’equilibrio della flora microbica presente nel latte, inibendo principalmente lo sviluppo dei fermenti lattici. In tal modo la cagliata (pasta filata formatasi per l’azione di fermenti) resta voluminosa, vischiosa, molle e ripiena di siero che non può gocciolare; inoltre si ha un abnorme sviluppo di coliformi che determinano una produzione di gas che la fa gonfiare producendo in essa numerosi buchi.
 resta voluminosa, vischiosa, molle e ripiena di siero che non può gocciolare; inoltre si ha un abnorme sviluppo di coliformi che determinano una produzione di gas che la fa gonfiare producendo in essa numerosi buchi.")
83
Anche in altre lavorazioni si hanno inconvenienti: nella produzione di burro, dove vengono alterati la formazione di acido e dell’aroma, di crema acida, di yogurt, di siero di latte. E’ da notare però che, nei centri di raccolta (con centinaia di vacche) il latte di un animale contenente antibiotico viene ampiamente diluito con altro latte genuino, privo di farmaci e la concentrazione di antibiotico può divenire talmente bassa e non più in grado di interferire nei processi di caseificazione, tranne che nella produzione di latti fermentati, dove è necessario esclusivamente l’utilizzo di latte indenne da farmaci.
 il latte di un animale contenente antibiotico viene ampiamente diluito con altro latte genuino, privo di farmaci e la concentrazione di antibiotico può divenire talmente bassa e non più in grado di interferire nei processi di caseificazione, tranne che nella produzione di latti fermentati, dove è necessario esclusivamente l’utilizzo di latte indenne da farmaci.")
Presentazioni simili



















