Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Estrazione e quantizzazione
degli acidi nucleici Pre-PCR Post-PCR Accettazione Refertazione

2
Area PCR GUANTI MONOUSO PUNTALI CON FILTRO CESTELLO PER GHIACCIO
TUBINI PCR

3
La diagnosi molecolare di una malattia genetica può essere effettuata:
direttamente, quando è nota la mutazione causativa indirettamente, quando: si conosce solo la localizzazione del locus (sito cromosomico) sul genoma umano l’indagine diretta non ha identificato la mutazione causativa
 sul genoma umano. l’indagine diretta non ha identificato la mutazione causativa.")
4
Analisi diretta Consiste nella ricerca diretta della/e mutazione/i
E’ particolarmente indicata nei casi in cui il gene da studiare non sia eccessivamente grande o in quei casi in cui siano presenti mutazioni ricorrenti La tecnica più idenea per l’identificazione diretta di una mutazione è la PCR

5
UN PO’ DI STORIA… Alla fine degli anni ’70, Kary Mullis si dedica alla ricerca sulla sintesi degli oligonucleotidi. Nel 1984, mette a punto la PCR (acronimo di Polymerase Chain Reaction), procedimento che consente di copiare (amplificare) esponenzialmente in vitro specifici tratti di DNA, moltiplicandolo miliardi di volte nel giro di poche ore.
, procedimento che consente di copiare (amplificare) esponenzialmente in vitro specifici tratti di DNA, moltiplicandolo miliardi di volte nel giro di poche ore.")
6
CARATTERISTICHE 5’ 3’ I primers utilizzati per la PCR sono definiti:
E’ una tecnica che consiste nell’amplificazione esponenziale in vitro di specifiche sequenze di DNA comprese tra le estremità 5’ di due specifici primer che fungono da innesco. I primers utilizzati per la PCR sono definiti: forward primer complementare al filamento di DNA 3'→5' reverse primer complementare al filamento di DNA 5'→3‘ La posizione dei primers delimita la regione di DNA da amplificare 5’ 3’

7
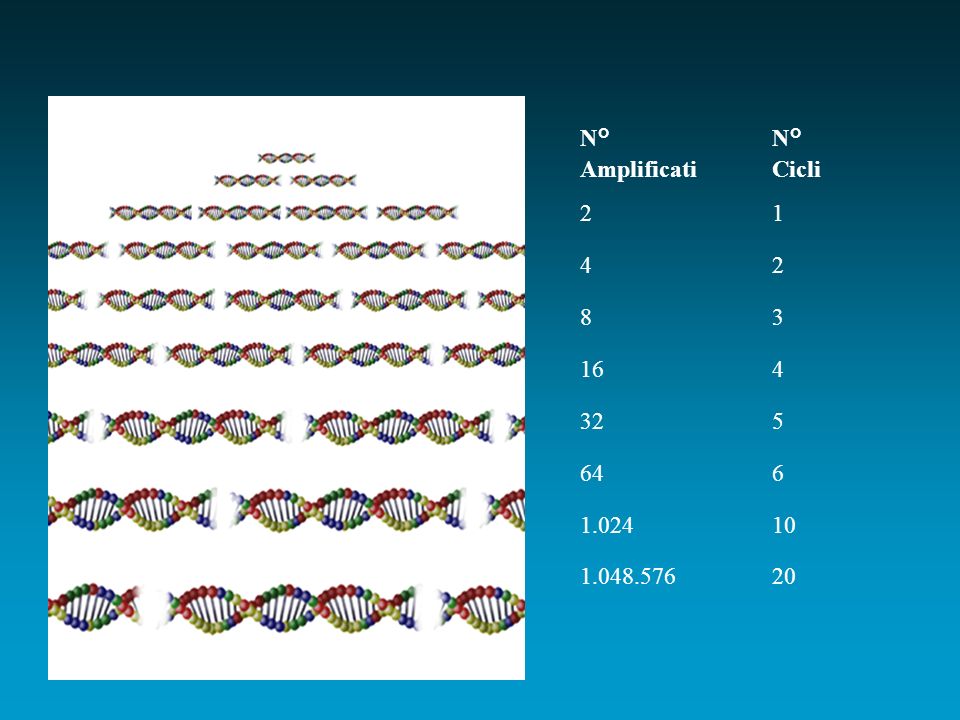
CARATTERISTICHE Può essere applicata solo quando è nota la sequenza nucleotidica di almeno un piccolo segmento di DNA su ogni lato della regione d’interesse (sequenza fiancheggiante). L’amplificazione si realizza con un processo ciclico in 3 tempi (denaturazione, appaiamento ed estensione). Consente di ottenere milioni di copie di una specifica sequenza di DNA anche a partire da una sua singola copia . L’amplificazione della sequenza bersaglio è uguale a 2n (entrambi i filamenti vengono copiati durante la PCR) ossia è esponenziale. Supposto che vi sia una sola copia del gene target prima dell’inizio dei cicli, dopo un ciclo vi saranno due copie, dopo due cicli vi saranno 4 copie…Dopo 20 cicli di PCR la sequenza bersaglio si sarà amplificata 220 volte pari a volte!!!
. L’amplificazione si realizza con un processo ciclico in 3 tempi (denaturazione, appaiamento ed estensione). Consente di ottenere milioni di copie di una specifica sequenza di DNA anche a partire da una sua singola copia . L’amplificazione della sequenza bersaglio è uguale a 2n (entrambi i filamenti vengono copiati durante la PCR) ossia è esponenziale. Supposto che vi sia una sola copia del gene target prima dell’inizio dei cicli, dopo un ciclo vi saranno due copie, dopo due cicli vi saranno 4 copie…Dopo 20 cicli di PCR la sequenza bersaglio si sarà amplificata 220 volte pari a volte!!!")
8
N° N° Amplificati Cicli
2 1 4 2 8 3 16 4 32 5 64 6
9
Applicazioni La PCR ha rivoluzionato il campo della diagnostica molecolare, fornendo nuovi e potenti strumenti per effettuare la diagnosi precoce di alterazioni genetiche anche a livello di singolo nucleotide. La PCR è oramai un mezzo insostituibile in diagnostica clinica per la diagnosi molecolare di: malattie genetiche ereditarie (identificazione di portatori, diagnosi prenatale) malattie genetiche acquisite (identificazione delle traslocazioni responsabili dell’insorgenza delle leucemie acute e croniche, analisi precoce di marcatori tumorali,) malattie sostenute da microrganismi patogeni e parassiti In campo forense e medico legale, la PCR si è imposta come strumento essenziale sul quale si basano analisi di paternità e perizie medico-legali ormai utilizzate comunemente a scopo probatorio nei tribunali di tutto il mondo.
 malattie genetiche acquisite (identificazione delle traslocazioni responsabili dell’insorgenza delle leucemie acute e croniche, analisi precoce di marcatori tumorali,) malattie sostenute da microrganismi patogeni e parassiti. In campo forense e medico legale, la PCR si è imposta come strumento essenziale sul quale si basano analisi di paternità e perizie medico-legali ormai utilizzate comunemente a scopo probatorio nei tribunali di tutto il mondo.")
10
I quattro deossiribonucleotidi trifosfato (dATP,dGTP,dCTP,dTTP)
Componenti della reazione di PCR Una DNA polimerasi termostabile MgCl2 aumenta la specificità della reazione favorendo l’appaiamento dei primer, la funzione della Taq polimerasi I quattro deossiribonucleotidi trifosfato (dATP,dGTP,dCTP,dTTP) Una coppia di primer o inneschi oligonucleotidici di sintesi (~20 nucleotidi) complementari alle regioni fiancheggianti la sequenza DNA target
 Una coppia di primer o inneschi oligonucleotidici. di sintesi (~20 nucleotidi) complementari alle regioni fiancheggianti la sequenza DNA target.")
11
Denaturazione termica
FASI DELLA METODICA Vi sono 3 step fondamentali in una PCR che vengono ripetuti per cicli all’interno di un thermalcycler. Denaturazione termica Appaiamento della coppia di primer alla porzione terminale della sequenza bersaglio: Estensione

12
1. DENATURAZIONE Avviene a 94-95°C
Apertura della doppia elica del DNA templato con formazione di due singoli filamenti (single stranded DNA o ssDNA) In questa fase tutte le reazioni enzimatiche si fermano (es. l’estensione del ciclo precedente)
 In questa fase tutte le reazioni enzimatiche si fermano (es. l’estensione del ciclo precedente)")
13
2. ANNEALING (APPAIAMENTO) O IBRIDIZZAZIONE DEI PRIMER
Avviene ad una temperatura compresa tra °C In questa fase, ciascuno dei due primer si lega con un legame ionico all’estremità 3’ della rispettiva catena a lui complementare. A questo punto, la DNA polimerasi può legarsi e cominciare a copiare il templato. Una volta formatosi un frammento di più basi, il legame ionico fra templato e primer diviene così forte da non andare più incontro a rotture. 3’ 5’ primer primer 5’ 3’

14
3. ESTENSIONE Nella fase di estensione (72°C) ha inizio e si completa la polimerizzazione di un filamento complementare a quello stampo a partire dal primer. Le basi (complementari al templato) vengono aggiunte all’estremità 3' del primer e la polimerizzazione avviene, ovviamente, in direzione 5’→3’. La sintesi ha luogo su entrambi i filamenti contemporameamente.
 ha inizio e si completa la polimerizzazione di un filamento complementare a quello stampo a partire dal primer. Le basi (complementari al templato) vengono aggiunte all’estremità 3 del primer e la polimerizzazione avviene, ovviamente, in direzione 5’→3’. La sintesi ha luogo su entrambi i filamenti contemporameamente.")
15
Conclusione del primo ciclo di PCR

16
Cinetica della PCR FASE ESPONENZIALE FASE LINEARE - FASE DI PLATEAU
reagenti in eccesso i prodotti si accumulano in modo costante reagenti si stanno consumando la reazione rallenta e i prodotti iniziano a degradarsi reagenti esauriti arresto della reazione Qual’è la modalità con la quale si accumula il prodotto in una reazione di PCR

17
Polymerase Chain Reaction
Log[DNA] N° cicli Esponenziale Lineare Plateau Prodotto variabile

18
TERMOCYCLER per PCR

19
Materiale di laboratorio per eseguire la PCR
Pipette Puntali con filtro Provette Porta-provette Cestello del ghiaccio guanti

20
REAGENTI PER PCR MIX STANDARD
Per preparare una PCR standard è necessario allestire una mix con: Acqua distillata autoclavata con cui portare a volume la mix Buffer 10x MgCl2 Soluzione equimolare dei quattro deossiribonucleotidi trifosfato (dNTP) La coppia di primer che ibridano le sequenze fiancheggianti bersaglio. Ciascun primer è un oligonucleotide costituito solitamente da un numero di nucleotidi variabile da 18 a 30 La Taq polimerasi DNA templato
 La coppia di primer che ibridano le sequenze fiancheggianti bersaglio. Ciascun primer è un oligonucleotide costituito solitamente da un numero di nucleotidi variabile da 18 a 30. La Taq polimerasi. DNA templato.")
21
MgCl2 Se presenti nella PCR mix a concentrazione ottimale (fra 0,5 a 2,5 mM) gli ioni magnesio: favoriscono l’appaiamento dei primer alle rispettive regioni fiancheggianti; favoriscono la funzione della Taq polimerasi; aumentano la specificità della reazione favorendo la formazione di prodotti di amplificazione specifici (amplificazione del tratto di DNA compreso fra i due primer). Concentrazione insufficiente = riduzione del numero di prodotti di amplificazione. Concentrazione eccessiva = inaccuratezza e aumento di prodotti aspecifici, conseguente all’aumentata frequenza di appaiamento dei primer con sequenze non omologhe .
. Concentrazione insufficiente = riduzione del numero di prodotti di amplificazione. Concentrazione eccessiva = inaccuratezza e aumento di prodotti aspecifici, conseguente all’aumentata frequenza di appaiamento dei primer con sequenze non omologhe .")
22
DEOSSIRIBONUCLEOTIDI TRIFOSFATO (dNTP)
I dNTP sono i mattoni necessari per costruire i nuovi filamenti di DNA. dATP o 2’-Deossiadenosina-5’-Trifosfato dGTP o 2’-Deossiguanosina-5’-Trifosfato dCTP o 2’-Deossicitidina-5’-Trifosfato dTTP o 2’Deossitimidina-5’-Trifosfato Un deossiribonucleotide è il monomero (o singola unità) di DNA. Ogni deossiribonucleotide è costituito da tre parti: una base azotata, uno zucchero deossiribosio ed uno o più gruppi fosfato. La base azotata è sempre legata al carbonio 1' del deossiribosio, che si distingue dal ribosio per la presenza di un protone sul carbonio 2' al posto di un gruppo —OH. Il gruppo fosfato si trova legato al carbonio 5' del deossiribosio. Quando i deossiribonucleotidi si polimerizzano a formare il DNA, il gruppo fosfato di un nucleotide si lega al carbonio 3' del successivo nucleotide tramite un legame fosfodiestere. I nuovi nucleotidi vengono sempre aggiunti al carbonio 3' dell'ultimo nucleotide, quindi la sintesi procede dall'estremità 5' alla 3'.
 di DNA. Ogni deossiribonucleotide è costituito da tre parti: una base azotata, uno zucchero deossiribosio ed uno o più gruppi fosfato. La base azotata è sempre legata al carbonio 1 del deossiribosio, che si distingue dal ribosio per la presenza di un protone sul carbonio 2 al posto di un gruppo —OH. Il gruppo fosfato si trova legato al carbonio 5 del deossiribosio. Quando i deossiribonucleotidi si polimerizzano a formare il DNA, il gruppo fosfato di un nucleotide si lega al carbonio 3 del successivo nucleotide tramite un legame fosfodiestere. I nuovi nucleotidi vengono sempre aggiunti al carbonio 3 dell ultimo nucleotide, quindi la sintesi procede dall estremità 5 alla 3 .")
23
DEOSSIRIBONUCLEOTIDI TRIFOSFATO (dNTP)
Nella Mix di reazione si aggiunge una miscela equimolare dei 4 nucleotidi trifosfati la cui concentrazione ottimale nella PCR varia in base alla lunghezza del tratto specifico da amplificare ed al numero di cicli di reazione impostati; quindi, va determinata sperimentalmente. Concentrazioni insufficienti riducono la resa della PCR. Concentrazioni eccessive inibiscono l’attività della Taq polimerasi.

24
PRIMER Sono prodotti in vitro per sintesi chimica ed hanno una lunghezza compresa fra 15 e 30 nucleotidi (primer più lunghi forniranno maggiore specificità). L’amplificazione di ogni sequenza target richiede la selezione e l’impiego di una specifica coppia di primer, la cui sequenza nucleotidica sia complementare a quella delle rispettive regioni fiancheggianti. Durante la PCR, ciascun primer ibridizza con la regione fiancheggiante complementare (annealing) e funge da innesco per la successiva reazione di estensione (polimerizzazione) catalizzata da una DNA polimerasi termostabile (Taq Polimerasi).
. L’amplificazione di ogni sequenza target richiede la selezione e l’impiego di una specifica coppia di primer, la cui sequenza nucleotidica sia complementare a quella delle rispettive regioni fiancheggianti. Durante la PCR, ciascun primer ibridizza con la regione fiancheggiante complementare (annealing) e funge da innesco per la successiva reazione di estensione (polimerizzazione) catalizzata da una DNA polimerasi termostabile (Taq Polimerasi).")
25
PRIMER La concentrazione nella PCR mix varia da 0,1 a 1 μM
Un eccesso di primer nella PCR mix provoca: formazione di prodotti di amplificazione “aspecifici”, riducendo la quantità di amplificato finale appaiamento dei primer tra di loro (dimeri) e non dei primer al tratto di DNA da amplificare, riducendo l’efficienza di amplificazione
 e non dei primer al tratto di DNA da amplificare, riducendo l’efficienza di amplificazione.")
26
Taq DNA polimerasi La DNA polimerasi catalizza la sintesi di DNA in direzione 5’→3’ a partire da un primer che funge da innesco. Inizialmente, le PCR venivano effettuate utilizzando polimerasi termolabili, che dovevano essere rimpiazzate ad ogni ciclo di amplificazione. Un significativo miglioramento della PCR si deve all’introduzione di una polimerasi termostabile, isolata da Thermus aquaticus, un batterio termofilo presente nelle fonti termali, detta Taq DNA polimerasi. Tale enzima ha un optimum a 72°C e resiste alle alte temperature usate negli step di denaturazione (95°C per oltre 40 min.), permettendo: un deciso miglioramento della specificità della reazione
, permettendo: un deciso miglioramento della specificità della reazione.")
27
Taq DNA polimerasi Le concentrazioni di Taq Polimerasi nella PCR mix variano da 0,25 a 2 Unità. Concentrazioni insufficienti causano una riduzione della resa di PCR Concentrazioni eccessive di enzima possono causare la sintesi di prodotti aspecifici

28
DNA TEMPLATO Può essere DNA genomico o cDNA.
Deve essere puro: evitare tutti i contaminanti chimici che interferiscono con l'attività della DNA polimerasi: EDTA chela gli ioni Mg2+, indispensabile per le polimerasi Sali che potrebbero formare complessi nel sito attivo dell'enzima ed inibirlo Fenolo Proteinasi K La quantità di templato da aggiungere alla PCR mix varia da 0.1 a 1μg, per un volume di reazione di 50 μl. Concentrazioni eccessive di DNA inibiscono l’attività della Taq polimerasi ed aumentano la formazione di prodotti di amplificazione aspecifici.

29
ELETTROFORESI SU GEL D’AGAROSIO
Una volta terminata la PCR e prima che il prodotto di PCR sia usato in ulteriori applicazioni, è necessario controllarlo mediante elettroforesi su gel d’agarosio per verificare se: La sequenza target è stata effettivamente amplificata. Il prodotto sia della giusta dimensione. E’ possibile che i primer oltre che ai siti desiderati ibridizzino anche ad altri tratti. In questo caso, sul gel si visualizzano più bande bande sulla stessa corsia.

30
ELETTROFORESI SU GEL D’AGAROSIO
L’elettroforesi è un processo elettrocinetico che consiste nella migrazione, sotto l’influenza di un campo elettrico, di molecole e particelle cariche in direzione del polo di carica opposta. Qualsiasi apparecchiatura per elettroforesi è composta da due componenti principali: un alimentatore ed una camera di separazione o cella elettroforetica correlata da due elettrodi. Per la separazione di campioni di DNA si adopera un apparato a configurazione orizzontale. Grazie alla presenza dei gruppi fosfato (PO4 3-), le molecole di DNA sono cariche negativamente e quindi migreranno verso il polo positivo (anodo) se sottoposte ad un campo elettrico.
, le molecole di DNA sono cariche negativamente e quindi migreranno verso il polo positivo (anodo) se sottoposte ad un campo elettrico.")
31
GEL D’AGAROSIO La separazione di molecole di DNA viene eseguita su gel d’agarosio, che funge da mezzo di supporto. Il gel d’agarosio è un polisaccaride lineare ed è uno dei costituenti dell’agar, una miscela di polisaccaridi isolati da alcune specie di alghe rosse. Viene generalmente utilizzato a concentrazioni dell’1-3%. Il gel viene prodotto sospendendo l’agarosio in polvere in un tampone acquoso (TBE 1x), portando la miscela ad ebollizione fino ad ottenere una soluzione limpida che viene versata in un apposito lettino e lasciata raffreddare a temperatura ambiente per formare un gel rigido. La misura dei pori del gel è funzione della concentrazione iniziale di agarosio: il diametro dei pori si riduce all’aumentare della concentrazione. Il gel viene posto all’interno della camera elettroforetica sommerso in una soluzione tampone (TBE 1x, pH 8.3, Tris, acido borico, EDTA) che ha la funzione di: favorire la conduzione della corrente che si genera quando fra i due elettrodi della camera viene applicata una differenza di potenziale; mantenere costante lo stato di ionizzazione delle molecole che devono essere separate (ogni variazione di pH provocherebbe una alterazione della carica totale modificando la mobilità, ossia la velocità di migrazione delle molecole da separare.
, portando la miscela ad ebollizione fino ad ottenere una soluzione limpida che viene versata in un apposito lettino e lasciata raffreddare a temperatura ambiente per formare un gel rigido. La misura dei pori del gel è funzione della concentrazione iniziale di agarosio: il diametro dei pori si riduce all’aumentare della concentrazione. Il gel viene posto all’interno della camera elettroforetica sommerso in una soluzione tampone (TBE 1x, pH 8.3, Tris, acido borico, EDTA) che ha la funzione di: favorire la conduzione della corrente che si genera quando fra i due elettrodi della camera viene applicata una differenza di potenziale; mantenere costante lo stato di ionizzazione delle molecole che devono essere separate (ogni variazione di pH provocherebbe una alterazione della carica totale modificando la mobilità, ossia la velocità di migrazione delle molecole da separare.")
32
Generalmente, si utilizza il blu di bromofenolo che, essendo provvisto di cariche negative, migrerà nella stessa direzione del DNA, ossia verso l’elettrodo positivo. Prima di essere caricato nel corrispettivo pozzetto del gel, ciascun campione viene mescolato con un marker colorato in modo da potere seguire la migrazione del DNA attraverso il gel.

33
VISUALIZZAZIONE CON ETIDIO BROMURO
Completata la “corsa” su gel d’agarosio ed avvenuta la seprazione dei vari frammenti di DNA di diversa lunghezza, è necessario procedere alla loro visualizzazione. A tale scopo, si utilizza il bromuro di etidio (EtBr), una sostanza che se irradiata da raggi UV ad una lunghezza d’onda di nm emette fluorescenza ( nm) di colore viola-arancio. Tale dye fluorescente viene aggiunto al gel d’agarosio ancora liquido prima che questo venga versato nell’apposito lettino per solidificare. Durante la migrazione del DNA attraverso il gel, il bromuro di etidio si intercala alle basi del DNA. Al trans-illuminatore nel gel i vari frammenti di DNA di lunghezza differente sono visibili sottoforma di bande fluorescenti presenti nella stessa corsia.
, una sostanza che se irradiata da raggi UV ad una lunghezza d’onda di nm emette fluorescenza ( nm) di colore viola-arancio. Tale dye fluorescente viene aggiunto al gel d’agarosio ancora liquido prima che questo venga versato nell’apposito lettino per solidificare. Durante la migrazione del DNA attraverso il gel, il bromuro di etidio si intercala alle basi del DNA. Al trans-illuminatore nel gel i vari frammenti di DNA di lunghezza differente sono visibili sottoforma di bande fluorescenti presenti nella stessa corsia.")
34
VISUALIZZAZIONE CON ETIDIO BROMURO

35
CONTAMINAZIONI La PCR è una tecnica molto sensibile.
Pertanto, è facilmente soggetta a contaminazioni da parte di molecole di DNA genomico proveniente da fonti diverse da quelle di interesse o da parte di prodotti di amplificazione (ampliconi) di precedenti PCR presenti in laboratorio. Il rischio consiste nell’amplificazione di DNA parassita (falsi positivi). Per tale ragione, è assolutamente necessario aggiungere ad ogni PCR dei controlli negativi (costituito da tutti i componenti della PCR mix eccetto il DNA templato).
 di precedenti PCR presenti in laboratorio. Il rischio consiste nell’amplificazione di DNA parassita (falsi positivi). Per tale ragione, è assolutamente necessario aggiungere ad ogni PCR dei controlli negativi (costituito da tutti i componenti della PCR mix eccetto il DNA templato).")
36
CONTROLLI DI QUALITA’ Controllo negativo 1: amplificazione di (H2O distillata) con i reagenti della reazione Controllo negativo 2 amplificazione del DNA di un soggetto sano Controllo negativo 3 amplificazione del DNA di un gene endogeno per valutare la qualità del campione Controllo positivo amplificazione del DNA di un soggetto con la mutazione

37
PREVENIRE LE CONTAMINAZIONI
Locali separati per estrazione acidi nucleici, conservazione degli acidi nucleici, preparazione PCR, amplificazione, elettroforesi su gel d’agarosio. Vetreria dedicata solo a PCR Guanti monouso Acqua deionizzata ed autoclavata Pipette automatiche con punte resistenti all’aerosol Preparazione della PCR eseguita sotto cappa a flusso laminare Decontaminazione periodica delle pipette automatiche con radiazioni UV

38
proteina p210 Controllo di qualità 3 2 5 6 8 3 1 4 7 CP CN 1 3 1 4 7 2
b3a2 b2a2 CN 1 proteina p210 3 1 4 7 2 5 6 8 Controllo di qualità 3 CP

39
Una variante della PCR è la RT-PCR
Consente di valutare l’espressione di determinati mRNA in una cellula o in un tessuto Estrazione di RNA totale Retro-trascrizione in cDNA (RNA DNA) Amplificazione con oligonucleotidi specifici 4. Analisi dei prodotti di amplificazione mediante elettroforesi su gel di agarosio
 Amplificazione con oligonucleotidi specifici. 4. Analisi dei prodotti di amplificazione mediante elettroforesi su gel di agarosio.")
40
Processo di retrotrascrizione
La retrotrascrizione (o trascrizione inversa) è, in biologia, la capacità da parte di particolari enzimi di sintetizzare una molecola di DNA a partire da RNA. L'enzima responsabile di questa caratteristica viene chiamato trascrittasi inversa, e deve il nome proprio al fatto che è in grado di compiere il passaggio inverso rispetto agli altri enzimi responsabili della trascrizione, che cioè sintetizzano RNA a partire da DNA, costituendo così una eccezione al dogma centrale della biologia molecolare. La trascrittasi inversa (chiamata anche "DNA polimerasi RNA-dipendente") è stata finora identificata nel genoma dei retrovirus (tra cui il virus dell'HIV), dai quali viene utilizzata per copiare l’informazione contenuta nel genoma retrovirale (che è costituito da RNA) in una molecola di DNA a doppio filamento che può così integrarsi (inserirsi) nel genoma della cellula ospite, dal quale può venire poi normalmente trascritto dando origine sia alle proteine virali che al genoma virale stesso. In questo modo il virus può rimanere in forma latente anche per molti anni. cDNA PCR
 è, in biologia, la capacità da parte di particolari enzimi di sintetizzare una molecola di DNA a partire da RNA. L enzima responsabile di questa caratteristica viene chiamato trascrittasi inversa, e deve il nome proprio al fatto che è in grado di compiere il passaggio inverso rispetto agli altri enzimi responsabili della trascrizione, che cioè sintetizzano RNA a partire da DNA, costituendo così una eccezione al dogma centrale della biologia molecolare. La trascrittasi inversa (chiamata anche DNA polimerasi RNA-dipendente ) è stata finora identificata nel genoma dei retrovirus (tra cui il virus dell HIV), dai quali viene utilizzata per copiare l’informazione contenuta nel genoma retrovirale (che è costituito da RNA) in una molecola di DNA a doppio filamento che può così integrarsi (inserirsi) nel genoma della cellula ospite, dal quale può venire poi normalmente trascritto dando origine sia alle proteine virali che al genoma virale stesso. In questo modo il virus può rimanere in forma latente anche per molti anni. cDNA. PCR.")
41
Reazione di retrotrascrizione (RT)
Perchè effettuiamo la retrotrascrizione: i punti di rottura su alcuni geni sono posizionati sugli esoni e quindi sono lontani L’RNA è abbastanza instabile e quindi convertirlo in CDNA lo rende più stabile La retrotrascrizione (o trascrizione inversa) è, in biologia, la capacità da parte di particolari enzimi di sintetizzare una molecola di DNA a partire da RNA. L'enzima responsabile di questa caratteristica viene chiamato trascrittasi inversa, e deve il nome proprio al fatto che è in grado di compiere il passaggio inverso rispetto agli altri enzimi responsabili della trascrizione, che cioè sintetizzano RNA a partire da DNA, costituendo così una eccezione al dogma centrale della biologia molecolare. La trascrittasi inversa (chiamata anche "DNA polimerasi RNA-dipendente") è stata finora identificata nel genoma dei retrovirus (tra cui il virus dell'HIV), dai quali viene utilizzata per copiare l’informazione contenuta nel genoma retrovirale (che è costituito da RNA) in una molecola di DNA a doppio filamento che può così integrarsi (inserirsi) nel genoma della cellula ospite, dal quale può venire poi normalmente trascritto dando origine sia alle proteine virali che al genoma virale stesso. In questo modo il virus può rimanere in forma latente anche per molti anni. La retrotrascrizione è utilizzata anche in biologia molecolare in esperimenti di ingegneria genetica a partire da mRNA purificato e porta alla sintesi di DNA complementare (cDNA), normalmente privo di introni che può venire utilizzato per il clonaggio e la manipolazione del gene corrispondente.
 è, in biologia, la capacità da parte di particolari enzimi di sintetizzare una molecola di DNA a partire da RNA. L enzima responsabile di questa caratteristica viene chiamato trascrittasi inversa, e deve il nome proprio al fatto che è in grado di compiere il passaggio inverso rispetto agli altri enzimi responsabili della trascrizione, che cioè sintetizzano RNA a partire da DNA, costituendo così una eccezione al dogma centrale della biologia molecolare. La trascrittasi inversa (chiamata anche DNA polimerasi RNA-dipendente ) è stata finora identificata nel genoma dei retrovirus (tra cui il virus dell HIV), dai quali viene utilizzata per copiare l’informazione contenuta nel genoma retrovirale (che è costituito da RNA) in una molecola di DNA a doppio filamento che può così integrarsi (inserirsi) nel genoma della cellula ospite, dal quale può venire poi normalmente trascritto dando origine sia alle proteine virali che al genoma virale stesso. In questo modo il virus può rimanere in forma latente anche per molti anni. La retrotrascrizione è utilizzata anche in biologia molecolare in esperimenti di ingegneria genetica a partire da mRNA purificato e porta alla sintesi di DNA complementare (cDNA), normalmente privo di introni che può venire utilizzato per il clonaggio e la manipolazione del gene corrispondente.")
42
PCR: vantaggi e svantaggi
Velocità e facilità d’uso In poche ore si possono ottenere milioni di copie della sequenza di DNA target Sensibilità Virtualmente è possibile utilizzare il DNA di una singola cellula come template Robustezza Amplificazione possibile anche utilizzando DNA di bassa qualità Necessario conoscere la sequenza del DNA da amplificare per sintetizzare i primers 4) Non tutte le taq polimerasi hanno attività di proofreading
 Non tutte le taq polimerasi hanno attività di proofreading.")
43
RT-PCR Plateau Log[DNA] Lineare Prodotto variabile Esponenziale
La RT-PCR non è una tecnica quantitativa Log[DNA] N° cicli Esponenziale Lineare Plateau Prodotto variabile Altri due svantaggi: La resa condizionata dalla disponibilità di reagenti ed il fatto che in questo modo non è una tecnicca quantitativa. La linearita’ della reazione esponenziale della PCR è limitata solo a pochi cicli. L’amplificazione nella fase esponenziale non è rilevabile con i sistemi tradizionali. Abbiamo quindi bisogno di un sistema che ci consenta di rilevare la produzione del prodotto di PCR nella fase esponenziale che rappresenta l’unica fase in ciui abbiamo certamente il 100% della reazione
![RT-PCR Plateau Log[DNA] Lineare Prodotto variabile Esponenziale](http://slideplayer.it/slide/10381785/33/images/43/RT-PCR+Plateau+Log%5BDNA%5D+Lineare+Prodotto+variabile+Esponenziale.jpg "La RT-PCR non è una tecnica quantitativa. Log[DNA] N° cicli. Esponenziale. Lineare. Plateau. Prodotto variabile. Altri due svantaggi: La resa condizionata dalla disponibilità di reagenti ed il fatto che in questo modo non è una tecnicca quantitativa. La linearita’ della reazione esponenziale della PCR è limitata solo a pochi cicli. L’amplificazione nella fase esponenziale non è rilevabile con i sistemi tradizionali. Abbiamo quindi bisogno di un sistema che ci consenta di rilevare la produzione del prodotto di PCR nella fase esponenziale che rappresenta l’unica fase in ciui abbiamo certamente il 100% della reazione.")
44
Limiti della visualizzazione su gel d’agarosio
Scarsa precisione Bassa sensibilità Bassa risoluzione Discriminazione bande con size marker Etidio bromuro non è quantitativo Inoltre ci sono dei limiti anche dal punto di vista della visualizzazione su gel.

45
Soluzioni Utilizzare i dati ottenuti durante la fase esponenziale
Il prodotto di PCR è proporzionale al template iniziale Questo è reso possibile mediante il rilevamento, di una fluorescenza, che è proporzionale al prodotto di PCR La fluorescenza, durante ogni ciclo di amplificazione, può essere rilevata utilizzando uno strumento quantitativo ma anche dei marcatori fluorescenti il cui accumulo segue la stessa cinetica della reazione di PCR Una soluzione a questo svantaggio è stata la possibilità di far evolvere la tecnologia della PCR in modo da poter utilizzare i dati ottenuti...

46
RT-PCR Plateau Log[DNA] Lineare Prodotto variabile Esponenziale
La RT-PCR non è una tecnica quantitativa Log[DNA] N° cicli Esponenziale Lineare Plateau Prodotto variabile Altri due svantaggi: La resa condizionata dalla disponibilità di reagenti ed il fatto che in questo modo non è una tecnicca quantitativa. La linearita’ della reazione esponenziale della PCR è limitata solo a pochi cicli. L’amplificazione nella fase esponenziale non è rilevabile con i sistemi tradizionali. Abbiamo quindi bisogno di un sistema che ci consenta di rilevare la produzione del prodotto di PCR nella fase esponenziale che rappresenta l’unica fase in ciui abbiamo certamente il 100% della reazione
![RT-PCR Plateau Log[DNA] Lineare Prodotto variabile Esponenziale](http://slideplayer.it/slide/10381785/33/images/46/RT-PCR+Plateau+Log%5BDNA%5D+Lineare+Prodotto+variabile+Esponenziale.jpg "La RT-PCR non è una tecnica quantitativa. Log[DNA] N° cicli. Esponenziale. Lineare. Plateau. Prodotto variabile. Altri due svantaggi: La resa condizionata dalla disponibilità di reagenti ed il fatto che in questo modo non è una tecnicca quantitativa. La linearita’ della reazione esponenziale della PCR è limitata solo a pochi cicli. L’amplificazione nella fase esponenziale non è rilevabile con i sistemi tradizionali. Abbiamo quindi bisogno di un sistema che ci consenta di rilevare la produzione del prodotto di PCR nella fase esponenziale che rappresenta l’unica fase in ciui abbiamo certamente il 100% della reazione.")
47
PERCHE’ QUANTIFICARE (1)
Molte applicazioni in medicina o in ricerca richiedono la quantificazione del numero di copie target del campione di partenza per es: - determinazione della carica virale nel sangue per la diagnosi di infezioni di HIV o HCV - l’analisi dell’espressione genica dai livelli di mRNA

48
PERCHE’ QUANTIFICARE (2)
DNA: alterazioni quantitative, dei geni mutati genomi estranei (microorganismi, OGM) DNA fetale (nel sangue materno aumenta nei casi di sofferenza del feto) DNA tumorale (aumenta nei soggetti affetti da neoplasie) mRNA: trascritti virali variazioni temporali variazioni individuali variazioni patologiche trascritti anomali (fusione, splicing) La regolazione dell’espressione genica varia in funzione del soggetto, patologia, sesso ecc. Durante la vita cellulare, sopravvivenza, crescita e differenzazione si riflettono in variazioni di espressione genica. Quantizzare i livelli di espressione genica è un punto determinante nella comprensione della funzione di un gene La quantificazione di un gene in linee cellulari sottoposte a trattamento farmacologico ci permette di studiarne la sua modulazione
 DNA fetale (nel sangue materno aumenta nei casi di sofferenza del feto) DNA tumorale (aumenta nei soggetti affetti da neoplasie) mRNA: trascritti virali. variazioni temporali. variazioni individuali. variazioni patologiche. trascritti anomali (fusione, splicing) La regolazione dell’espressione genica varia in funzione del soggetto, patologia, sesso ecc. Durante la vita cellulare, sopravvivenza, crescita e differenzazione si riflettono in variazioni di espressione genica. Quantizzare i livelli di espressione genica è un punto determinante nella comprensione della funzione di un gene. La quantificazione di un gene in linee cellulari sottoposte a trattamento farmacologico ci permette di studiarne la sua modulazione.")
49
PCR Real Time PCR Real Time permette contemporaneamente
- l’amplificazione - il rilevamento dell’amplificato La PCR Real Time misura l’amplificazione in tempo reale durante la fase esponenziale della reazione Durante ogni ciclo di amplificazione viene rilevata la fluorescenza del campione proporzionale al prodotto di PCR La fluorescenza si genera durante la PCR per effetto di reazioni chimiche come il legame di molecole fluorescenti Analisi del prodotto di PCR non su gel di agarosio ma mediante valutazione della fluorescenza tramite software gestito da un computer

50
PCR Convenzionale vs. PCR Real-Time
Divergenza !!! rilevazione tradizionale Rilevazione Real-Time PCR

51
Ct Principio base della Real-Time PCR Sample
threshold Si determina QUANDO viene generato il segnale piuttosto che l‘intensità del segnale stesso (analisi all‘end –point) Ct = threshold cycle: Il numero di cicli al quale il prodotto di PCR supera la soglia di rilevabilità rilevazione tradizionale
 Ct = threshold cycle: Il numero di cicli al quale il prodotto di PCR supera la soglia di rilevabilità. rilevazione. tradizionale.")
52
Chimiche fluorescenti
per PCR Real-Time La fluorescenza si genera durante la PCR per effetto di diverse possibili reazioni chimiche Le chimiche principali sono basate sia sul legame di coloranti fluorescenti che si intercalano nella doppia elica di DNA, come il SYBR Green, sia sull'ibridazione di sonde specifiche.

53
SYBR Green

54
SYBR Green: principio Utilizza una molecola fluorescente non specifica che si lega al solco minore del DNA

55
SYBR Green All’inizio del processo di amplificazione, la miscela di reazione contiene DNA denaturato, primers e la molecola fluorescente

56
SYBR Green Dopo l’annealing dei primers, si legano poche molecole fluorescenti alla doppia elica.

57
SYBR Green Durante l’elongazione si verifica un aumento di fluorescenza che corrisponde all’ aumento del numero di copie dell’amplicone

58
SYBR green Metodica semplice Non costosa Non-specifica
La molecola fluorescente si lega in maniera aspecifica a tutte le doppie eliche. È necessario ottimizzare la metodica per evitare la formazione di prodotti aspecifici

59
TaqMan

60
TaqMan La Real-Time PCR si può realizzare mediante l’impiego:
coloranti intercalanti ( es. SYBR green), che si legano in maniera aspecifica a tutto il DNA sonde ad ibridazione specifiche per il frammento di interesse, marcate con molecole fluorescenti Esistono diversi tipi di sonde: Dual-labeled (come le sonde TaqMan) Molecular beacons Scorpion Sonde FRET (Fluorescence Resonance Energy Transfer)
, che si legano. in maniera aspecifica a tutto il DNA. sonde ad ibridazione specifiche per il frammento di interesse, marcate con molecole fluorescenti. Esistono diversi tipi di sonde: Dual-labeled (come le sonde TaqMan) Molecular beacons. Scorpion. Sonde FRET (Fluorescence Resonance Energy Transfer)")
61
Sonde TaqMan La sonda di tipo TaqMan è un oligonucleotide che, come i primers della PCR, viene disegnato per essere complementare alla sequenza bersaglio da amplificare Primer 3’ 5’ R Q La sonda è disegnata in modo da ibridarsi all’interno del frammento amplificato nella reazione di PCR

62
Reporter-Quencher R Q 5’ 3’
5’ REPORTER (R): fluorocromo ad alta energia che emette fluorescenza 3’ QUENCHER (Q): fluorocromo a bassa energia che spegne la fluorescenza del reporter 5’ 3’ R Q Se R e Q si trovano vicini, Q spegne l'effetto di R perchè i fotoni di R vengono assorbiti da Q
: fluorocromo ad alta energia che emette fluorescenza. 3’ QUENCHER (Q): fluorocromo a bassa energia che spegne la fluorescenza del reporter. 5’ 3’ R. Q. Se R e Q si trovano vicini, Q spegne l effetto di R perchè i fotoni di R vengono assorbiti da Q.")
63
attività 5’>3’ esonucleasica
Real-Time PCR: attività 5’>3’ esonucleasica R Q 3’ 5’ Attività esonucleasica di correzione

64
Curve di amplificazione
Cicli di amplificazione Fluorescenza Plot lineare Per ogni campione si ottiene una curva di amplificazione il cui CT(=Threshold Cycle) è inversamente proporzionale alla quantità di template iniziale
 è inversamente proporzionale alla quantità di template iniziale.")
65
Sonde FRET (Fluorescence Resonance Energy Transfer)
donatore accettore Simili alle sonde TaqMan perché si legano al DNA bersaglio e vengono idrolizzate, ci sono però due sonde ognuna marcata con un solo fluorocromo (accettore e donatore) Quando le sonde non sono legate alle sequenze target il segnale fluorescente proveniente dall'accettore non è rilevato Durante lo step di annealing PCR, entrambe le sonde FRET ibridizzano alle sequenze target: ciò avvicina il fluoroforo donatore all'accettore permettendo il trasferimento di energia tra i due fluorofori e la produzione di un segnale fluorescente da parte dell'accettore che viene rilevato
 Quando le sonde non sono legate alle sequenze target il segnale fluorescente proveniente dall accettore non è rilevato Durante lo step di annealing PCR, entrambe le sonde FRET ibridizzano alle sequenze target: ciò avvicina il fluoroforo donatore all accettore permettendo il trasferimento di energia tra i due fluorofori e la produzione di un segnale fluorescente da parte dell accettore che viene rilevato")
66
Molecular Beacons I "molecular beacons" contengono un fluoroforo e un quencher alle estremità opposte di un oligonucleotide, che sono disegnate in modo da essere complementari tra loro formando una struttura stem-loop Il loop è complementare ad una sequenza all'interno del prodotto amplificato La vicinanza del quencher al reporter fluorescente impedisce l’emissione di fluorescenza quencher fluoroforo
![]()
67
Molecular Beacons Durante lo step di annealing PCR, la sonda ibridizza alla sua sequenza target: ciò separa il colorante fluorescente dal reporter, producendo un segnale fluorescente Amplicon FRET EXCITATIONE ANNEALING La quantità di fluorescenza prodotta ad ogni ciclo, o dopo la PCR, dipende dalla quantità di prodotto specifico in quel dato momento A differenza delle sonde TaqMan, le molecular beacons non vengono distrutte durante la reazione di amplificazione per cui possono reibridizzarsi durante il successivo ciclo
![]()
68
Gli Scorpions® sono primers di PCR con una coda “stem-loop” che contiene un fluoroforo e un quencher. La coda “stem-loop” è separate dalla sequenza del primer di PCR da un “PCR blocker”, una modificazione chimica che impedisce alla Taq DNA polymerase di copiare la sequenza dello Scorpions®. Questo impedisce l’apertura non specifica del loop che porterebbe all’emissione di fluorescenza aspecifica. Gli Scorpions® differiscono dalle metodiche di detection tradizionali dal momento che i loro meccanismo di “probing” è intramolecolare. Durante la PCR, i primers Scorpions® vengono allungati per produrre prodotti di PCR. Durante la fase di annealing la sequenza sonda nella coda dello Scorpion scivola indietro per ibridizzare con la sequenza target contenuta nel prodotto di PCR neoformato. Dal momento che la coda dello Scorpions® e il prodotto di PCR sono ora parte dello stesso strand, l’interazione è intramolecolare, cineticamente favorevole e altamente efficiente.

69
Real-Time PCR: applicazioni
Quantificazione virale Quantificazione dell’espressione genica Efficacia della terapia farmacologica Misura dei danni al DNA Controllo di qualità e validazione dei saggi Detenzione dei patogeni Controllo degli OGM Genotyping MRD

70
MRD (Minimal Residual Disease):
Quota residua di cellule neoplastiche non eradicate dalla terapia farmacologica Tali elementi neoplastici, presenti ad un livello inferiore alla capacità di rilevazione delle metodiche convenzionali, sono in grado di espandersi e dare origine alla recidiva

71
TEST GENETICI: DEFINIZIONE
Insieme di analisi che vengono effettuate sul DNA, RNA,cromosomi, proteine metaboliti che consentono di evidenziare alterazioni dovute ad una patologia genetica, ereditabile o meno. Essi si distinguono in test clinici e di ricerca

72
TEST GENETICI: CLINICI
Forniscono al paziente ed alla famiglia una risposta utile per l’inquadramento diagnostico, la prevenzione o un trattamento terapeutico I laboratori che effettuano test clinici devono seguire delle procedure approvate e standardizzate

73
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
TEST GENETICI: CLINICI Test diagnostici: per confermare o escludere una malattia genetica nota o sospetta in un individuo sintomatico Test predittivi: per individui asintomatici in una famiglia con una storia di una patologia genetica. Tali test sono indicati nel caso in cui si possa ridurre la morbidità o mortalità della malattia. Test di portatore:per identificare individui con una storia familiare o anche a gruppi etnici con un elevato numero di portatori di uno specifico allele patologico Test prenatali, effettuati in gravidanza per determinare lo stato di salute del feto.Tali test si effettuano solo se si conoscono in anticipo le mutazioni legate alla patologia nella famiglia

74
TEST GENETICI di ricerca
Tali test hanno invece lo scopo principale è la migliore comprensione di un disordine genetico o lo sviluppo di una nuova tecnologia diagnostica

75
TEST CLINICI DIRETTI: quando si conosce la mutazione causativa della malattia INDIRETTI: si conosce solo la localizzazione del locus (sito cromosomico) sul genoma umano l’indagine diretta non ha identificato la mutazione causativa DIRETTO = il campione di un consultando è testato per individuare un’alterazione patogenetica in un determinato gene. Dà informazioni riguardanti quell’individuo. INDIRETTO = marcatori polimorfici sono utilizzati in studi familiari per valutare se il consultando ha ereditato l’allele che verosimilmente veicola l’alterazione patogenetica. Si esegue su una famiglia e dà informazioni su quale dei due alleli di un segmento cromosomale segrega nei vari membri della famiglia.
 sul genoma umano. l’indagine diretta non ha identificato la mutazione causativa. DIRETTO = il campione di un consultando è testato per individuare un’alterazione patogenetica in un determinato gene. Dà informazioni riguardanti quell’individuo. INDIRETTO = marcatori polimorfici sono utilizzati in studi familiari per valutare se il consultando ha ereditato l’allele che verosimilmente veicola l’alterazione patogenetica. Si esegue su una famiglia e dà informazioni su quale dei due alleli di un segmento cromosomale segrega nei vari membri della famiglia.")
76
I TEST DIRETTI si eseguono per diagnosticare
Malattie ereditarie: alterazioni qualitative o quantitative del materiale genetico a livello delle cellule germinali trasmissione alla prole Malattie acquisite: alterazioni qualitative o quantitative del materiale genetico a livello delle cellule somatiche - Malattie ereditarie: attraverso la rilevazione di alterazioni qualitative o quantitative di materiale genetico endogeno (mutazioni del genoma umano) a livello di tutti i tessuti (DNA) o di singoli tessuti (RNA) - Malattie acquisite (a livello somatico): tumori solidi, leucemie, etc. attraverso la rilevazione di alterazioni di materiale genetico (DNA) endogeno a livello di tessuti, organi, cellule -attraverso la rilevazione di modificazioni dell’espressione genica: analisi di mRNA qualitative e/o quantitative non viene trasmessa alla prole
 a livello di tutti i tessuti (DNA) o di singoli tessuti (RNA) - Malattie acquisite (a livello somatico): tumori solidi, leucemie, etc. attraverso la rilevazione di alterazioni di materiale genetico (DNA) endogeno a livello di tessuti, organi, cellule. -attraverso la rilevazione di modificazioni dell’espressione genica: analisi di mRNA qualitative e/o quantitative. non viene trasmessa alla prole.")
77
I TEST DIRETTI si eseguono per diagnosticare
FIBROSI CISTICA TALASSEMIA DISTROFIA DI DUCHENNE/BECKER EMOFILIA ATROFIA MUSCOLO-SPINALE Malattie ereditarie Malattie acquisite MALATTIE EMOLINFOPROLIFERATIVE MALATTIE INFETTIVE Tumori

78
MALATTIE EREDITARIE Sono autosomiche se trasmesse dai cromosomi autosomici (dal cromosoma 1 al cromosoma 22) Sono legate al cromosoma X (x linked) se trasmesse dal cromosoma X Mutazioni dominanti: condizioni in un individuo eterozigote, in cui il l’allele mutato si esprime fenotipicamente. si parla di dominanza di un allele su di un altro quando, in un individuo eterozigote, solo l'allele dominante si esprime, ossia influenza il fenotipo si parla di recessività relativamente ad un allele quando, in un individuo eterozigote, il carattere indotto dall'allele in questione non si manifesta fenotipicamente Mutazioni recessive:condizioni in un individuo eterozigote in cui l’allele mutato non si manifesta fenotipicamente.
 se trasmesse dal cromosoma X. Mutazioni dominanti: condizioni in un individuo eterozigote, in cui il l’allele mutato si esprime fenotipicamente. si parla di dominanza di un allele su di un altro quando, in un individuo eterozigote, solo l allele dominante si esprime, ossia influenza il fenotipo. si parla di recessività relativamente ad un allele quando, in un individuo eterozigote, il carattere indotto dall allele in questione non si manifesta fenotipicamente. Mutazioni recessive:condizioni in un individuo eterozigote in cui l’allele mutato non si manifesta fenotipicamente.")
79
I test INDIRETTI solo per diagnosticare
Malattie ereditarie si conosce la localizzazione del locus (sito cromosomico) sul genoma umano Analisi dei polimorfismi del DNA Polimorfismo genetico:variazione genetica nella popolazione caratterizzata da sostituzioni, delezioni o inserzioni di basi nel DNA MALATTIE EREDITARIE Analisi dei polimorfismi:Si parla di polimorfismo genetico quando una variazione genetica ha una prevalenza maggiore dell'1% nella popolazione. La variazione genetica può essere determinata da sostituzioni, delezioni o inserzioni di basi nel DNA e può riguardare regioni codificanti e regioni non codificanti. attraverso analisi di linkage genetico (marcatori co-segreganti con la lesione genica) a livello del DNA: Analisi dei polimorfismi di sequenza (RFLP e SNP) Analisi dei polimorfismi di lunghezza (VNTR e STR) Può riguardare regioni codificanti Marcatori che vengono trasmessi con la lesione genetica
 sul genoma umano. Analisi dei polimorfismi del DNA. Polimorfismo genetico:variazione genetica nella popolazione caratterizzata da sostituzioni, delezioni o inserzioni di basi nel DNA. MALATTIE EREDITARIE. Analisi dei polimorfismi:Si parla di polimorfismo genetico quando una variazione genetica ha una prevalenza maggiore dell 1% nella popolazione. La variazione genetica può essere determinata da sostituzioni, delezioni o inserzioni di basi nel DNA e può riguardare regioni codificanti e regioni non codificanti. attraverso analisi di linkage genetico (marcatori co-segreganti con la lesione genica) a livello del DNA: Analisi dei polimorfismi di sequenza (RFLP e SNP) Analisi dei polimorfismi di lunghezza (VNTR e STR) Può riguardare regioni codificanti. Marcatori che vengono trasmessi con la lesione genetica.")
80
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Analisi mediante linkage disequilibrium Termine genetico con cui si indica la presenza di associazione statistica tra alleli specifici che costituiscono un determinato aplotipo il linkage disequilibrium è maggiore in popolazioni omogenee, cioè originate da un nucleo di individui fondatori come le popolazioni sarda o finlandese Individua regioni cromosomiche in cui si collocano i geni responsabili di una data malattia Analisi molecolare delle varianti alleliche in pazienti apparentemente non imparentati. Il linkage disequilibrium (LD) è un termine dell'ambito genetico che indica la presenza di associazione statistica tra specifici alleli relativi a due o più loci, che costituiscono di solito un particolare aplotipo (Con il termine aplotipo si definisce la combinazione di varianti alleliche lungo un cromosoma o segmento cromosomico contenente loci in linkage disequilibrium, cioè strettamente associati tra di loro[1], e che in genere vengono ereditati insieme) ancestrale, diffuso nella popolazione in cui è rilevato perché trasmesso lungo la discendenza da un comune progenitore. Per questo motivo il linkage disequilibrium è maggiore in popolazioni omogenee, cioè originate da un nucleo di individui fondatori come le popolazioni sarda o finlandese. Infatti le migrazioni recenti generano substruttura ed eterogeneità genetica e aplotipi differenti associati allo stesso tratto tendono ad abbassare i valori di significatività. Il linkage disequilibrium è un importante strumento per individuare regioni cromosomiche di limitata ampiezza in cui si collocano i geni per una data malattia (mappaggio ad alta risoluzione) e si avvale dell'analisi molecolare di varianti alleliche (per lo più di SNPs o STRs) che costituiscono aplotipi in pazienti tra loro apparentemente non imparentati. Infatti è prevedibile che pazienti che hanno ereditato lo stesso segmento cromosomico, definito dal medesimo aplotipo, abbiano ereditato anche la stessa mutazione in esso contenuto. Risale agli anni '80, il primo gene mappato con questo approccio: quello della fibrosi cistica in 7q31.2. I pazienti che hanno ereditato lo stesso segmento cromosomico, ereditano anche la stessa mutazione in esso contenuto. Risale agli anni '80, il primo gene mappato con questo approccio: quello della fibrosi cistica in 7q31.2.
 è un termine dell ambito genetico che indica la presenza di associazione statistica tra specifici alleli relativi a due o più loci, che costituiscono di solito un particolare aplotipo (Con il termine aplotipo si definisce la combinazione di varianti alleliche lungo un cromosoma o segmento cromosomico contenente loci in linkage disequilibrium, cioè strettamente associati tra di loro[1], e che in genere vengono ereditati insieme) ancestrale, diffuso nella popolazione in cui è rilevato perché trasmesso lungo la discendenza da un comune progenitore. Per questo motivo il linkage disequilibrium è maggiore in popolazioni omogenee, cioè originate da un nucleo di individui fondatori come le popolazioni sarda o finlandese. Infatti le migrazioni recenti generano substruttura ed eterogeneità genetica e aplotipi differenti associati allo stesso tratto tendono ad abbassare i valori di significatività. Il linkage disequilibrium è un importante strumento per individuare regioni cromosomiche di limitata ampiezza in cui si collocano i geni per una data malattia (mappaggio ad alta risoluzione) e si avvale dell analisi molecolare di varianti alleliche (per lo più di SNPs o STRs) che costituiscono aplotipi in pazienti tra loro apparentemente non imparentati. Infatti è prevedibile che pazienti che hanno ereditato lo stesso segmento cromosomico, definito dal medesimo aplotipo, abbiano ereditato anche la stessa mutazione in esso contenuto. Risale agli anni 80, il primo gene mappato con questo approccio: quello della fibrosi cistica in 7q31.2. I pazienti che hanno ereditato lo stesso segmento cromosomico, ereditano. anche la stessa mutazione in esso contenuto. Risale agli anni 80, il primo gene mappato con questo approccio: quello della fibrosi cistica in 7q31.2.")
81
Analisi molecolare di polimorfismi
di sequenza (SNPs-single nucleotide polimorfism, RFLP-restriction fragment lenght polymorphism) di lunghezza (VNTR Variable number tandem repeats, STR-single tandem repeats)
 di lunghezza (VNTR Variable number tandem repeats, STR-single tandem repeats)")
82
VALIDITA’ DEI TEST GENETICI
La maggior parte dei test genetici non è in grado di identificare tutte le mutazioni patogenetiche dipende da: eterogeneità genetica:quando mutazioni in loci genetici diversi possono avere lo stesso effetto fenotipico eterogeneità allelica :quando mutazioni diverse sono a carico dello stesso locus La sensibilità di un test genetico dipende da: eterogeneità allelica eterogeneità genica della malattia in esame La maggior parte dei test genetici non è in grado di identificare tutte le mutazioni patogenetiche (p.e. la sensibilità diagnostica quando si ricercano macrodelezioni nel caso della distrofia muscolare di Duchenne/Beker è di circa il 65%). Ciononostante, quando una mutazione è ritrovata in una famiglia, essa può essere rivelata nella stessa famiglia con una sensibilità del 100%.
. Ciononostante, quando una mutazione è ritrovata in una famiglia, essa può essere rivelata nella stessa famiglia con una sensibilità del 100%.")
83
TEST GENETICI Affetti Non affetti Validità diagnostica + al test VP FP
- al test FN VN Valutazione della validità diagnostica dei test genetici Sensibilità del test = veri positivi/totale affetti = VP/(VP + FN) Specificità del test = veri negativi/totale sani = VN/(VN +FP)
 Specificità del test = veri negativi/totale sani = VN/(VN +FP)")
Presentazioni simili



















>")