Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
DISORDINI X-LINKED Sono circa 500 i geni mappati sul cromosoma X, il 70% di questi sono associati a malattia.

2
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
DISORDINI X-LINKED Gene sul cromosoma X I maschi, emizigoti per il cromosoma X, sono più spesso affetti Nelle femmine, entrambe le copie devono essere mutate

3
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
DISORDINI X-LINKED Distrofia muscolare di Duchenne/Becker Emofilia A e B

4
DISTROFIA MUSCOLARE DI DUCHENNE/BECKER (DMD/BMD)
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy DISTROFIA MUSCOLARE DI DUCHENNE/BECKER (DMD/BMD) Miopatia ereditaria degenerativa, dovuta ad un difetto del gene che sintetizza la distrofina una proteina che si lega alla membrana delle fibre muscolari e ne preserva l’integrità. Incidenza: 1/3500 nati maschi 1/30000 per la forma Becker
 Miopatia ereditaria degenerativa, dovuta ad un difetto del gene che sintetizza la distrofina una proteina che si lega alla membrana delle fibre muscolari e ne preserva l’integrità. Incidenza: 1/3500 nati maschi. 1/30000 per la forma Becker.")
5
DISTROFIA MUSCOLARE DI DUCHENNE/BECKER (DMD/BMD)
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy DISTROFIA MUSCOLARE DI DUCHENNE/BECKER (DMD/BMD) La malattia è trasmessa con carattere recessivo X-linked I maschi che veicolano mutazioni nel gene DMD sono affetti Le femmine portatrici sono sane Le mutazioni recessive devono coinvolgere i due alleli per manifestarsi clinicamente
 La malattia è trasmessa con carattere recessivo X-linked. I maschi che veicolano mutazioni nel gene DMD sono affetti. Le femmine portatrici sono sane. Le mutazioni recessive devono coinvolgere i due alleli per manifestarsi clinicamente.")
6
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
DUCHENNE o BECKER? DMD - Duchenne Muscular Dystrophy Forma grave. Assenza totale o forte riduzione della sintesi di distrofina BMD - Becker Muscular Dystrophy Fenotipo più lieve. Distrofina ridotta o strutturalmente alterata

7
Caratteristiche cliniche DMD
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy Caratteristiche cliniche DMD Miopatia degenerativa progressiva Insorgenza nella forma grave tra i 2 e 5 anni Infiltrazione nel muscolo di tessuto connettivo e adiposo Pseudoipertrofia muscolare Progressiva necrosi muscolare Complicanze polmonari e/o cardiache portano al decesso Riduzione o assenza della distrofina esordio caratterizzato da andatura anserina, frequenti cadute e difficoltà a salire le scale atrofia progressiva prima dei muscoli prossimali degli arti inferiori e poi prossimale degli arti superiori e di seguito sono colpiti i muscoli respiratori cardiomiopatia nel 95% dei casi negli ultimi anni di vita Progressivamente il muscolo viene sostituito da tessuto adiposo e connettivo

8
CARATTERISTICHE CLINICHE Distrofia di Becker (BMD)
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy CARATTERISTICHE CLINICHE Distrofia di Becker (BMD) Forma allelica della DMD Meno grave Insorgenza tra 12 e 80 anni Mutazioni del gene della distrofina che conservano parzialmente la funzionalità della proteina forma allelica: variazione della sequenza dello stesso gene
 Forma allelica della DMD. Meno grave. Insorgenza tra 12 e 80 anni. Mutazioni del gene della distrofina che conservano parzialmente la funzionalità della proteina. forma allelica: variazione della sequenza dello stesso gene.")
9
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
DISTROFINA - IL GENE Localizzato sul braccio corto del cromosoma X Lungo più di 3000 kb, pari a circa 1,5 % dell’intero cromosoma X Costituito da 79 esoni

10
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
La proteina distrofina espressa soprattutto nel muscolo scheletrico e in piccole quantità nel cervello

11
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Complesso delle glicoproteine associate alla distrofina (DAG) citoscheletro I-band Muscolo Fascio di Fibra muscolare Proteina Membrana cellulare IL RUOLO DELLA DISTROFINA. IL COMPLESSO PROTEICO CUI PARTECIPA LA DISTROFINA COLLEGA IL CITOSCHELETRO (regola l’impalcaura della cellula) ALLA MATRICE EXTRACELLULARE E FORNISCE SUPPORTO ALLA MEMBRANA DURANTE I CICLI DI CONTRAZIONE E RILASSAMENTO. ALTERAZIONI GENETICHE DELLA DISTROFINA CONDUCONO A DANNI ALLA MEMBRANA E A MORTE DELLE cellule muscolari.
 citoscheletro. I-band. Muscolo. Fascio di Fibra muscolare. Proteina. Membrana cellulare. IL RUOLO DELLA DISTROFINA. IL COMPLESSO PROTEICO CUI PARTECIPA LA DISTROFINA COLLEGA IL CITOSCHELETRO (regola l’impalcaura della cellula) ALLA MATRICE EXTRACELLULARE E FORNISCE SUPPORTO ALLA MEMBRANA DURANTE I CICLI DI CONTRAZIONE E RILASSAMENTO. ALTERAZIONI GENETICHE DELLA DISTROFINA CONDUCONO A DANNI ALLA MEMBRANA E A MORTE DELLE cellule muscolari.")
12
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Ruolo della distrofina La distrofina funge da ponte tra l’apparato contrattile intracellulare e la matrice extra-cellulare, sottoposta a forte stress meccanico durante la contrazione muscolare. In assenza di distrofina, anche tutte le proteine dell’apparato contrattile diminuiscono; pertanto la mancanza di distrofina produce un difetto al supporto strutturale della membrana, rendendola più suscettibile a rotture durante l’attività contrattile. Nuovi studi sul ruolo della distrofina:oltre al ruolo meccanico, regola l’attività di altri geni coinvolti nella fibrosi e nello stress ossidativo Recentemente è stato scoperto che accanto a questo ruolo strutturale, la distrofina ha anche una funzione più sofisticata, quella di controllare l’attività di altri geni che hanno un ruolo rilevante nella sviluppo della malattia. Si tratta di geni che contengono le informazioni per piccoli RNA (microRNA) capaci di controllare in modo molto preciso alcuni fenomeni rilevanti nella patogenesi della distrofia, quali lo stress ossidativo e la fibrosi
 capaci di controllare in modo molto preciso alcuni fenomeni rilevanti nella patogenesi della distrofia, quali lo stress ossidativo e la fibrosi.")
13
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
BASI GENETICHE Circa il 65% delle mutazioni DMD e BMD sono grosse delezioni del gene che portano alla totale assenza o a ridottissime quantità di distrofina delezioni con scivolamento di lettura del codice DMD delezioni con lettura corretta BMD

14
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Tipi di mutazioni del gene della distrofina Macrodelezioni ~ 65% Macroduplicazioni ~ 5-10% Mutazioni puntiformi, micro-inserzioni e -delezioni ~ 25-30%

15
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Diagnosi di BMD/ DMD 1. biochimica creatina fosfochinasi sierica elevata (CPK) 2. istopatologica biopsia muscolare -colorazione per la distrofina 3. Analisi del DNA screening di delezioni comuni o mutazioni note in famiglia Aumenta in caso di infarto, aritmie, traumi distrofie muscolari, neoplasie.tale enzima viene dosato con saggi enzimatici
 2. istopatologica. biopsia muscolare -colorazione per la distrofina. 3. Analisi del DNA. screening di delezioni comuni o mutazioni note in famiglia. Aumenta in caso di infarto, aritmie, traumi distrofie muscolari, neoplasie.tale enzima viene dosato con saggi enzimatici.")
16
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Indagine Biochimica Determinazione dei livelli sierici della componente muscolare MM della CPK valori di rifierimento (v.n. uomo 350 UI/l - donna 250 UI/l) Maschi % individui affetti Concentrazione CK sierica DMD 100% > 10 volte la norma BMD > 5 volte la norma Maschi % individui affetti Concentrazione CPK sierica DMD 100% >10 volte la norma BMD >5 volte la norma L’incremento dei livelli di CPK è evidente fin dalla nascita e raggiunge il suo picco intorno ai tre anni per poi attenuarsi.Questa cinetica riflette l’andamento progressivo della malattia nel senso che all’inizio c’è un attiva necrosi con tentativi di rigenerazione muscolare e poi gradualmente si cede alla scomparsa delle fibre muscolari sostituite da tessuto fibro-adiposo.
 Maschi. % individui affetti. Concentrazione CK sierica. DMD. 100% > 10 volte la norma. BMD. > 5 volte la norma. Maschi. % individui affetti. Concentrazione CPK sierica. DMD. 100% >10 volte la norma. BMD. >5 volte la norma. L’incremento dei livelli di CPK è evidente fin dalla nascita e raggiunge il suo picco intorno ai tre anni per poi attenuarsi.Questa cinetica riflette l’andamento progressivo della malattia nel senso che all’inizio c’è un attiva necrosi con tentativi di rigenerazione muscolare e poi gradualmente si cede alla scomparsa delle fibre muscolari sostituite da tessuto fibro-adiposo.")
17
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Indagine Biochimica Determinazione dei livelli sierici della componente muscolare MM della CPK valori di rifierimento (v.n. uomo 350 UI/l - donna 250 UI/l) Maschi % individui affetti Concentrazione CK sierica DMD 100% > 10 volte la norma BMD > 5 volte la norma Femmine portatrici % individui affetti Concentrazione CPK sierica DMD circa 50% 2-10 volte la norma BMD circa 30% L’incremento dei livelli di CPK è evidente fin dalla nascita e raggiunge il suo picco intorno ai tre anni per poi attenuarsi.Questa cinetica riflette l’andamento progressivo della malattia nel senso che all’inizio c’è un attiva necrosi con tentativi di rigenerazione muscolare e poi gradualmente si cede alla scomparsa delle fibre muscolari sostituite da tessuto fibro-adiposo. Nelle fasi di attiva necrosi muscolare possono aumentare i livelli di LDH, transaminasi e aldolasi
 Maschi. % individui affetti. Concentrazione CK sierica. DMD. 100% > 10 volte la norma. BMD. > 5 volte la norma. Femmine portatrici. % individui affetti. Concentrazione CPK sierica. DMD. circa 50% 2-10 volte la norma. BMD. circa 30% L’incremento dei livelli di CPK è evidente fin dalla nascita e raggiunge il suo picco intorno ai tre anni per poi attenuarsi.Questa cinetica riflette l’andamento progressivo della malattia nel senso che all’inizio c’è un attiva necrosi con tentativi di rigenerazione muscolare e poi gradualmente si cede alla scomparsa delle fibre muscolari sostituite da tessuto fibro-adiposo. Nelle fasi di attiva necrosi muscolare possono aumentare i livelli di LDH, transaminasi e aldolasi.")
18
X X DIAGNOSI ISTOLOGICA Delezioni genomiche (assenza di distrofina)
colorazione della distrofina sul contorno delle fibre muscolari di soggetto sano Distrofia di Duchenne: assenza di distrofina Distrofia di Becker: ridotta colorazione delle fibre Tronca o normale (BMD) X Delezioni genomiche (assenza di distrofina) X
 X. Delezioni genomiche (assenza di distrofina) X.")
19
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Tecniche Molecolari per la diagnosi della DMD Western blot per l’espressione della distrofina da biopsia muscolare di pazienti con BMD o DMD effettuato su biopsie muscolari di pazienti affetti

20
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Analisi del DNA File 1-2: distrofia di Becker Fila 3: normale espressione di distrofina File 4-5: distrofia di Duchenne Western blot per distrofina da biopsia muscolare di pazienti con BMD o DMD

21
Cos’e’ un Western Blot? Una tecnica in cui le proteine sono separate mediante elettroforesi su gel e successivamente trasferite su un supporto (membrana o filtro). Successivamente la specifica proteina di cui stiamo andando a valutare l’espressione viene identificata mediante la sua reazione con un anticorpo specifico.
. Successivamente la specifica proteina di cui stiamo andando a valutare l’espressione viene identificata mediante la sua reazione con un anticorpo specifico.")
22
Fasi di un Western Blot Prima fase: elettroforesi su gel.
(Le proteine del campione vengono separate su un gel in base alle loro dimensioni) Seconda fase: trasferimento su membrana. (Le proteine nel gel sono poi trasferite su una membrana di nitrocellulosa mediante un campo elettrico) Terza fase: saturazione o “blocking”. (La saturazione e’ usata per prevenire le interazioni non specifiche tra l’anticorpo e la membrana)
 Seconda fase: trasferimento su membrana. (Le proteine nel gel sono poi trasferite su una membrana di nitrocellulosa mediante un campo elettrico) Terza fase: saturazione o blocking . (La saturazione e’ usata per prevenire le interazioni non specifiche tra l’anticorpo e la membrana)")
23
Fasi di un Western Blot Quarta fase: legame dell’anticorpo primario.
(L’anticorpo riconosce la proteina specifica immobilizzata sulla membrana) Quinta fase: legame dell’anticorpo secondario. (L’anticorpo secondario, coniugato a un enzima (AP o HRP), riconosce specificamente l’anticorpo primario, gia’ legato alla proteina sulla membrana) Sesta fase: rivelazione o “detection”. (L’enzima coniugato all’anticorpo secondario scinde un substrato che, in corrispondenza della proteina specifica, sviluppa precipitato colorato o chemioluminescente)
 Quinta fase: legame dell’anticorpo secondario. (L’anticorpo secondario, coniugato a un enzima (AP o HRP), riconosce specificamente l’anticorpo primario, gia’ legato alla proteina sulla membrana) Sesta fase: rivelazione o detection . (L’enzima coniugato all’anticorpo secondario scinde un substrato che, in corrispondenza della proteina specifica, sviluppa precipitato colorato o chemioluminescente)")
24
PRIMA FASE: denaturazione e CORSA ELETTROFORETICA

25
Western blot: seconda fase Immobilizzazione e trasferimento
Elettroblotting Apparato di trasferimento Il gel e’ messo tra strati di carta da filtro con la membrana a diretto contatto col gel sul lato verso l’elettrodo positivo Viene applicato un campo elettrico e le proteine migrano fuori dal gel verso l’elettrodo positivo e si legano alla membrana Fatto a 4°C per evitare surriscaldamento, decomposizione del tampone e degradazione delle proteine

26
Seconda fase Immobilizzazione e trasferimento
membrana

27
Western blot: seconda fase Immobilizzazione e trasferimento
Trasferimento dal catodo (-) all’ anodo (+) Spugnette 3 fogli di carta da filtro imbevuti di tampone di trasferimento Gel Membrana
 all’ anodo (+) Spugnette. 3 fogli di carta da filtro imbevuti di tampone di trasferimento. Gel. Membrana.")
28
Apparato per il trasferimento “Semi-dry”

29
Western blot: terza fase saturazione o “blocking”
Per saturare i siti idrofobici liberi sulla membrana Per prevenire il legame dell’anticorpo primario alla membrana stessa Latte scremato o Albumina di Siero Bovino (BSA) reducing background
 reducing background.")
30
Western blot: quarta fase incubazione con anticorpo primario
L’anticorpo primario riconosce la proteina di interesse e non lega le altre proteine immobilizzate sulla membrana

31
Western blot: quarta fase incubazione con anticorpo primario

32
Western blot: quinta fase Incubazione con anticorpo secondario

33
Western blot: sesta fase rivelazione o “detection”
Fosfatasi alcalina (AP) o perossidasi del rafano (HRP) Conversione di un substrato colorimetrico in un precipitato colorato Substrati Chemioluminescenti Emettono luce se convertiti dall’enzima Possono essere visualizzati su lastre radiografiche Marcatura radioattiva
 o perossidasi del rafano (HRP) Conversione di un substrato colorimetrico in un precipitato colorato. Substrati Chemioluminescenti. Emettono luce se convertiti dall’enzima. Possono essere visualizzati su lastre radiografiche. Marcatura radioattiva.")
34
HRP Western blot substrato luce Anticorpo primario Anticorpo secondario Rivelazione Il substrato metabolizzato dalla perossidasi (HRP) emette luce pg proteina
 emette luce. pg proteina.")
35
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Analisi diretta: Macro-delezioni del gene della distrofina Sono localizzate preferenzialmente in due zone del gene: la deletion rich region prossimale (esoni 1-20) e la deletion rich region distale (esoni 45-53); le mutazioni out of frame sono responsabili più frequentemente del fenotipo Duchenne le mutazioni in frame sono responsabili più frequentemente del fenotipo Becker
 e la deletion rich region distale (esoni 45-53); le mutazioni out of frame sono responsabili più frequentemente del fenotipo Duchenne. le mutazioni in frame sono responsabili più frequentemente del fenotipo Becker.")
36
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Esoni del gene DMD …………………… … 53 Deletion rich prossimale Deletion rich distale

37
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Genetica Molecolare Analisi del DNA su leucociti del sangue Linee guida del gruppo collaborativo europeo per la diagnosi Molecolare della Distrofia DMB/DMD MPLA (multiple ligation probe amplification), analizza tutti gli esoni del gene della distrofina in contemporanea (delezioni e duplicazioni) Questo approccio consente di evidenziare circa il 98% delle delezioni del gene della distrofina
, analizza tutti. gli esoni del gene della distrofina in contemporanea. (delezioni e duplicazioni) Questo approccio consente di evidenziare circa il 98% delle. delezioni del gene della distrofina.")
38
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
MPLA Multiplex ligation-dependent probe amplification (MLPA)[1] is a variation of the multiplex polymerase chain reaction that permits multiple targets to be amplified with only a single primer pair.[1] Each probe consists of two oligonucleotides which recognise adjacent target sites on the DNA. One probe oligonucleotide contains the sequence recognised by the forward primer, the other the sequence recognised by the reverse primer. Only when both probe oligonucleotides are hybridised to their respective targets, can they be ligated into a complete probe. The advantage of splitting the probe into two parts is that only the ligated oligonucleotides, but not the unbound probe oligonucleotides, are amplified. the forward primer used for probe amplification is fluorescently labeled, each amplicon generates a fluorescent peak which can be detected by a capillary sequencer. Poiche solo le sonde legate vengono amplificate in maniera esponenziale durante la reazione di PCR, il numero di prodotti di PCR legati rappresenta una misura diretta delle sequenze target nel campione esaminato
[1] is a variation of the multiplex polymerase chain reaction that permits multiple targets to be amplified with only a single primer pair.[1] Each probe consists of two oligonucleotides which recognise adjacent target sites on the DNA. One probe oligonucleotide contains the sequence recognised by the forward primer, the other the sequence recognised by the reverse primer. Only when both probe oligonucleotides are hybridised to their respective targets, can they be ligated into a complete probe. The advantage of splitting the probe into two parts is that only the ligated oligonucleotides, but not the unbound probe oligonucleotides, are amplified. the forward primer used for probe amplification is fluorescently labeled, each amplicon generates a fluorescent peak which can be detected by a capillary sequencer. Poiche solo le sonde legate vengono amplificate in maniera esponenziale durante la reazione di PCR, il numero di prodotti di PCR legati rappresenta una misura diretta delle sequenze target nel campione. esaminato.")
39
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Quando le condizioni di amplificazione sono ottimizzate è possibile valutare i prodotti dell’amplificazione direttamente su gel d’agarosio Elettroforesi su gel di agarosio dei prodotti di amplificazione mediante MPLA: quando le condizioni di amplificazione sono ottimizzate è possibile valutare i prodotti dell’amplificazione direttamente su gel d’agarosio :

40
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
ELETTROFORESI CAPILLARE Migrazione dei campioni all’interno di un capillare di silice collegato a due elettrodi Strumenti automatizzati per l’analisi di frammenti e l’analisi di sequenze Utilizzo per l’amplificazione di un primer fluorescente Vantaggi Velocità di corsa Analisi di ridotte quantità di campione Alto potere risolutivo Alternativamente i prodotti della MPLA possono essere visualizzati medinate elettroforesi capillare che è un’evoluzione dell’elettroforesi classica su agarosio. Una volta screenate le delezioni o duplicazioni per multiplex , esse vengono visualizzate con elettroforesi capillare.

41
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Principio Una piccola quantità di campione viene caricata a livello dell’anodo del capillare in silice all’interno di un campione appropriato. Grazie all’applicazione di una ddp, le molecole migrano con differenti veocità lungo il capillare.

42
Diagnosi DMD mediante Elettroforesi capillare
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy Diagnosi DMD mediante Elettroforesi capillare

43
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Tipi di mutazione del gene della distrofina Macrodelezioni ~ 65% Macroduplicazioni ~ 5-10% Mutazioni puntiformi, micro-inserzioni e -delezioni ~ 25-30%

44
DHPLC Denaturing High Performance Liquid Chromatography
La tecnica, sviluppata nel laboratorio del Prof. Cavalli Sforza alla Stanford University (USA), permette la rilevazione di variazioni nella sequenza del DNA, e si basa sulla differente velocità di eluizione in una colonna cromatografica per gli eteroduplex e gli omoduplex. In particolare, in condizioni parzialmente denaturanti e sotto un diretto controllo della temperatura, permette di discriminare all’interno di prodotti eterogenei di PCR, molecole di DNA eteroduplex rispetto alle molecole omoduplex. Questo perchè le variazioni di sequenza creano una popolazione mista di eteroduplex ed omoduplex durante il re-annealing di DNA wild type e mutato. E’ una tecnica relativamente recente che sfrutta i principi della cromatografia liquida ad alta risoluzione (HPLC) per la separazione e l’analisi degli acidi nucleici.
, permette la rilevazione di variazioni nella sequenza del DNA, e si basa sulla differente velocità di eluizione in una colonna cromatografica per gli eteroduplex e gli omoduplex. In particolare, in condizioni parzialmente denaturanti e sotto un diretto controllo della temperatura, permette di discriminare all’interno di prodotti eterogenei di PCR, molecole di DNA eteroduplex rispetto alle molecole omoduplex. Questo perchè le variazioni di sequenza creano una popolazione mista di eteroduplex ed omoduplex durante il re-annealing di DNA wild type e mutato. E’ una tecnica relativamente recente che sfrutta i principi. della cromatografia liquida ad alta risoluzione (HPLC) per la separazione e l’analisi degli acidi nucleici.")
45
Principio del metodo: Fase stazionaria: copolimero non polare idrofobico (polistirene-divinilbenzene) Fase mobile: 1) Tri-Etil Ammonio Acetato (TEAA), molecola anfipatica, che fungendo da ponte (accoppiante ionico), permette alle molecole di DNA (cariche negativamente) di interagire con la matrice 2) Acetonitrile (ACN), solvente organico 3) Acqua Opportune condizioni di gradiente di fase mobile, a concentrazione crescente di solvente organico (ACN), permettono prima l’adsorbimento e poi l’eluizione dei frammenti di acido nucleico in funzione della lunghezza (condizioni non denaturanti) e/o in funzione della sequenza (condizioni parzialmente denaturanti). Il principio di separazione cromatografica dei frammenti di DNA da analizzare è quello della cromatografia liquida a scambio ionico in fase inversa costituita da una fase stazionaria e da una fase mobile. La fase stazionaria consiste in una colonna riempita con una matrice non porosa del copolimero polistirene-divinilbenzene (PS-DVB). Le sfere sono alchilate con catene C-18 che formano singoli legami C-C. Per natura, le sfere (diametro 2 µm) sono elettrostaticamente neutre e idrofobiche e non reagiscono con gli acidi nucleici (in rosso nella figura). La fase mobile consiste in una combinazione di buffers (TEAA e acetonitrile a varie concentrazioni) che eluisce il DNA trattenuto dalle sfere dalla colonna cromatografica. Il trimetilammonioacetato (TEAA) agisce come “molecola ponte” per permettere l’adesione degli acidi nucleici alle sfere. Questa molecola ha una porzione idrofobica ed una porzione carica positivamente. La carica positiva del TEAA interagisce con la carica negativa dei gruppi fosfato degli acidi nucleici, mentre i gruppi idrofobici interagiscono con la catene C-18 delle sfere di PS-DVB. I campioni da analizzare sono amplificati normalmente come da protocolli standard di PCR ed eluiti ad una o più temperature stabilite dette “temperature di melting” ottenute mediante l’applicazione dell’ algoritmo di Stanford, che calcola la temperatura alla quale effettuare la corsa cromatografica che deve essere sufficiente per la parziale denaturazione dell’eteroduplex eventualmente presente (in presenza di una mutazione), ma non in grado di denaturare l’omoduplex. 4 reservoir Canale A: Buffer A HPLC grade (soluzione acquosa, 0.1M TEAA, pH 7.0) Canale B: Buffer B HPLC grade (soluzione acquosa, 0.1M TEAA, 25% ACN, pH 7.0) Canale C: Soluzione D HPLC grade (soluzione acquosa 75% ACN) Canale D: Soluzione di lavaggio della siringa (soluzione acquosa 8% ACN) I Buffer A e B sono soluzioni di lavoro che costituiscono la fase mobile. La percentuale di Buffer A e B varia durante nel corso dell’analisi per assicurare il gradiente di fase mobile a concentrazione di ACN progressivamente crescente.
 Tri-Etil Ammonio Acetato (TEAA), molecola anfipatica, che fungendo da ponte (accoppiante ionico), permette alle molecole di DNA (cariche negativamente) di interagire con la matrice. 2) Acetonitrile (ACN), solvente organico. 3) Acqua. Opportune condizioni di gradiente di fase mobile, a concentrazione crescente di. solvente organico (ACN), permettono. prima l’adsorbimento e poi l’eluizione. dei frammenti di acido nucleico. in funzione della lunghezza. (condizioni non denaturanti) e/o. in funzione della sequenza (condizioni parzialmente denaturanti). Il principio di separazione cromatografica dei frammenti di DNA da analizzare è quello della cromatografia liquida a scambio ionico in fase inversa costituita da una fase stazionaria e da una fase mobile. La fase stazionaria consiste in una colonna riempita con una matrice non porosa del copolimero polistirene-divinilbenzene (PS-DVB). Le sfere sono alchilate con catene C-18 che formano singoli legami C-C. Per natura, le sfere (diametro 2 µm) sono elettrostaticamente neutre e idrofobiche e non reagiscono con gli acidi nucleici (in rosso nella figura). La fase mobile consiste in una combinazione di buffers (TEAA e acetonitrile a varie concentrazioni) che eluisce il DNA trattenuto dalle sfere dalla colonna cromatografica. Il trimetilammonioacetato (TEAA) agisce come molecola ponte per permettere l’adesione degli acidi nucleici alle sfere. Questa molecola ha una porzione idrofobica ed una porzione carica positivamente. La carica positiva del TEAA interagisce con la carica negativa dei gruppi fosfato degli acidi nucleici, mentre i gruppi idrofobici interagiscono con la catene C-18 delle sfere di PS-DVB. I campioni da analizzare sono amplificati normalmente come da protocolli standard di PCR ed eluiti ad una o più temperature stabilite dette temperature di melting ottenute mediante l’applicazione dell’ algoritmo di Stanford, che calcola la temperatura alla quale effettuare la corsa cromatografica che deve essere sufficiente per la parziale denaturazione dell’eteroduplex eventualmente presente (in presenza di una mutazione), ma non in grado di denaturare l’omoduplex. 4 reservoir. Canale A: Buffer A HPLC grade (soluzione acquosa, 0.1M TEAA, pH 7.0) Canale B: Buffer B HPLC grade (soluzione acquosa, 0.1M TEAA, 25% ACN, pH 7.0) Canale C: Soluzione D HPLC grade (soluzione acquosa 75% ACN) Canale D: Soluzione di lavaggio della siringa (soluzione acquosa 8% ACN) I Buffer A e B sono soluzioni di lavoro che costituiscono la fase mobile. La percentuale di Buffer A e B varia durante nel corso dell’analisi per assicurare il gradiente di fase mobile a concentrazione di ACN progressivamente crescente.")
46
Caratteristiche La separazione dei frammenti di DNA in base alla diversa dimensione e alla diversa conformazione è permessa dal diverso adsorbimento e ripartizione tra la fase stazionaria (matrice) e la fase mobile (buffer). Procedura rapida completamente automatizzata, strumentazione costosa ma con bassi costi di analisi, Non è possibile localizzare e identificare precisamente la mutazione, utile per screening su grandi popolazioni.
 e la fase mobile (buffer). Procedura rapida completamente automatizzata, strumentazione costosa ma con bassi costi di analisi, Non è possibile localizzare e identificare precisamente la mutazione, utile per screening su grandi popolazioni.")
47
Applicazioni del DHPLC
Permette di separare ssDNA e dsDNA operando in tre diverse condizioni a seconda delle finalità desiderate: - non denaturanti, ~ 50°C (sizing dsDNA, STR) - parzialmente denaturanti, > 50°C (mutation detection, SNP) - totalmente denaturanti, > 75°C (sintesi e purificazione di oligo) Il DHPLC è caratterizzato da una elevata versatilità applicativa, infatti permette di separare ssDNA e dsDNA operando in tre diverse condizioni a seconda delle finalità desiderate: non denaturanti, ~ 50°C (sizing dsDNA, purificazione, STR, applicazioni Q-RT-PCR) parzialmente denaturanti, > 50°C (mutation detection, SNP) totalmente denaturanti, > 75°C (oligonucleotidi, RNA, purificazione)
 - parzialmente denaturanti, > 50°C (mutation detection, SNP) - totalmente denaturanti, > 75°C (sintesi e purificazione di oligo) Il DHPLC è caratterizzato da una elevata versatilità applicativa, infatti permette di separare ssDNA e dsDNA operando in tre diverse condizioni a seconda delle finalità desiderate: non denaturanti, ~ 50°C (sizing dsDNA, purificazione, STR, applicazioni Q-RT-PCR) parzialmente denaturanti, > 50°C (mutation detection, SNP) totalmente denaturanti, > 75°C (oligonucleotidi, RNA, purificazione)")
48
SIZING Condizioni non denaturanti Analisi di frammenti dsDNA
Il tempo di ritenzione è direttamente proporzionale alla lunghezza dei frammenti SIZING: determinazione del peso molecolare

49
A B bp 587 458 434 298 174 102 80 IL DHPLC permettee di effettuare una elevata risoluzione dei frammenti dsDNA Risoluzione delle bande del plasmide pUC18 digerito con l’enzima di restrizione HaeIII Con l’elettroforesi su gel di agarosio 0.8% non si riescono a distinguere i frammenti di 257 e 267 bp che co-migrano Con il sistema WAVE® si riescono a separare 9 frammenti

50
Analisi di mutazione e’ un metodo temperatura dipendente
basato sulla separazione cromatografica di molecole di DNA ds contenenti basi non appaiate (eteroduplex)
")
51
Applicazione del DHPLC nella DMD
Esempio di femmina con mutazione puntiforme ex 59 Esempio di femmina eterozigote con delezione ex 67

52
Limiti del DHPLC La metodica del DHPLC non consente l’identificazione della posizione della mutazione e tantomeno la sua tipologia, quindi per i campioni che mostrano profili di eluizione alterati rispetto ai wild-type è previsto sempre il successivo sequenziamento diretto.

53
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy
Trattamento e terapie Attualmente non esistono terapie. Tutti i trattamenti mirano al controllo dei sintomi e a migliorare la qualità della vita

54
TERAPIA GENICA PER DMD: cosa si è fatto e cosa si sta facendo
March 3, 2203: Single Gene 6: Duchenne Muscular Dystrophy TERAPIA GENICA PER DMD: cosa si è fatto e cosa si sta facendo Terapia genica mediante l’utilizzo di un vettore che trasporta una mini-distrofina (solo una parte della sequenza codificante). Il ruolo del vettore virale è quello di veicolare porzioni geniche per ripristinare il corretto schema di lettura nel DNA delle cellule muscolari del paziente e stimolare la produzione di nuova distrofina. Difficoltà delle’approccio: il gene responsabile, quello della distrofina, è molto grosso e quindi difficile da “impacchettare” nei vettori virali attualmente disponibili per il trasferimento nelle cellule dei malati Altra strategia usando l’RNA rispristinando la produzione di una distrofina più corta ma funzionale. Identificazione, come conseguenza del danno muscolare di microRNA, che rilasciati in circolo inibiscono l’esperessione genica. L’analisi di questi piccoli RNA potrebbe essere adoperata come un marker biologico per diagnosticare la malattia e misurare l’efficacia dei trattamenti terapeutici. La terapia genica (sostituzione del gene difettoso con quello funzionante) è difficilmente applicabile alla Dmd, perché il gene responsabile, quello della distrofina, è molto grosso e quindi difficile da “impacchettare” nei vettori virali attualmente disponibili per il trasferimento nelle cellule dei malati. Negli ultimi anni abbiamo sperimentato una nuova strategia che consiste nella correzione del difetto genico a livello dell’Rna: questa è la molecola a partire dalla quale si producono le proteine e che, grazie a un processo di “taglia e cuci”, nella sua forma matura ha una dimensione ridotta rispetto al Dna. Qualche anno fa avevamo dimostrato l’efficacia di questa strategia nel ripristinare la produzione di una distrofina più corta ma funzionale nel topo distrofico. Scopo di questo progetto è migliorare l’efficienza di produzione della distrofina in vista di una possibile terapia genica della Dmd nell’uomo. Infine, abbiamo identificato che, come conseguenza del danno muscolare, alcune molecole - piccoli Rna - che inibiscono l’espressione genica vengono rilasciati in circolo: intendiamo ora analizzare se questi piccoli Rna possano essere utilizzati come marcatori biologici per diagnosticare la malattia e misurare l’efficacia dei trattamenti terapeutici.
. Il ruolo del vettore virale è quello di veicolare porzioni geniche per ripristinare il corretto schema di lettura nel DNA delle cellule muscolari del paziente e stimolare la produzione di nuova distrofina. Difficoltà delle’approccio: il gene responsabile, quello della distrofina, è molto grosso e quindi difficile da impacchettare nei vettori virali attualmente disponibili per il trasferimento nelle cellule dei malati. Altra strategia usando l’RNA rispristinando la produzione di una distrofina più corta ma funzionale. Identificazione, come conseguenza del danno muscolare di microRNA, che rilasciati in circolo inibiscono l’esperessione genica. L’analisi di questi piccoli RNA potrebbe essere adoperata come un marker biologico per diagnosticare la malattia e misurare l’efficacia dei trattamenti terapeutici. La terapia genica (sostituzione del gene difettoso con quello funzionante) è difficilmente applicabile alla Dmd, perché il gene responsabile, quello della distrofina, è molto grosso e quindi difficile da impacchettare nei vettori virali attualmente disponibili per il trasferimento nelle cellule dei malati. Negli ultimi anni abbiamo sperimentato una nuova strategia che consiste nella correzione del difetto genico a livello dell’Rna: questa è la molecola a partire dalla quale si producono le proteine e che, grazie a un processo di taglia e cuci , nella sua forma matura ha una dimensione ridotta rispetto al Dna. Qualche anno fa avevamo dimostrato l’efficacia di questa strategia nel ripristinare la produzione di una distrofina più corta ma funzionale nel topo distrofico. Scopo di questo progetto è migliorare l’efficienza di produzione della distrofina in vista di una possibile terapia genica della Dmd nell’uomo. Infine, abbiamo identificato che, come conseguenza del danno muscolare, alcune molecole - piccoli Rna - che inibiscono l’espressione genica vengono rilasciati in circolo: intendiamo ora analizzare se questi piccoli Rna possano essere utilizzati come marcatori biologici per diagnosticare la malattia e misurare l’efficacia dei trattamenti terapeutici.")
55
Fibrosi cistica La Fibrosi cistica, detta anche mucoviscidosi e’ un malattia genetica autosomica recessiva. E’ una malattia poco conosciuta anche se si tratta della più comune fra le malattie genetiche mortali nelle popolazioni di origine caucasica. Frequenza:1:200-1:6000 nati

56
Fibrosi cistica Nella sua forma più grave, la FC colpisce diversi organi, tra cui pancreas, polmoni, fegato, intestino Questa patologia si caratterizza per un'anomalia nel trasporto del cloro nella membrana delle cellule delle ghiandole a secrezione esterna. Di conseguenza queste ghiandole secernono un muco denso e vischioso e quindi poco scorrevole. Negli organi interessati, le secrezioni mucose, essendo anormalmente viscide, determinano un'ostruzione dei dotti principali, provocando l'insorgenza di gran parte delle manifestazioni cliniche tipiche della malattia, come la comparsa di infezioni polmonari ricorrenti, di insufficienza pancreatica, di steatorrea, di stati di malnutrizione, di cirrosi epatica, di ostruzione intestinale e di infertilità maschile; è una malattia molto grave. Il termine fibrosi cistica deriva dal tessuto cicatriziale che si forma nel pancreas

57
PATOFISIOLOGIA (I) Funzionamento anomalo della proteina CFTR (cystic fibrosis transmembrane conductance) che normalmente presiede ad alcune funzioni di trasporto di sali e di difesa contro le infezioni. Questo difetto comporta che le secrezioni degli organi siano dense e poco scorrevoli ristagnando ed occludendo dotti e canali, con danno progressivo degli organi interessati.
 che normalmente presiede ad alcune funzioni di trasporto di sali e di difesa contro le infezioni. Questo difetto comporta che le secrezioni degli organi siano dense e poco scorrevoli ristagnando ed occludendo dotti e canali, con danno progressivo degli organi interessati.")
58
PATOFISIOLOGIA (II) Il pancreas si atrofizza e non libera i suoi enzimi digestivi (ad es. la tripsina), l’intestino si occlude,il fegato trattiene la bile, i bronchi si ostruiscono e si infettano Progressivamente compare il danno polmonare con infezioni ripetute fino all’infezione cronica che determina un’insufficienza respiratoria che è la causa principale di morte
, l’intestino si occlude,il fegato trattiene la bile, i bronchi si ostruiscono e si infettano. Progressivamente compare il danno polmonare con infezioni ripetute fino all’infezione cronica che determina un’insufficienza respiratoria che è la causa principale di morte.")
59
PATOFISIOLOGIA (III) Un tempo era considerata una malattia mortale dell’infanzia. Oggi il 50% dei pazienti supera I anni. Questo dato dipende da diversi fattori come la diagnosi precoce, il miglioramento delle cure,la diagnosi sempre più frequente di forme lievi. Il trapianto di polmone , diventato più frequente solo negli ultimi 10 anni, rappresenta un fattore decisivo per il prolungamento dell’aspettativa di vita nelle forme classiche gravi.

60
Screening neonatale per FC: razionale
1982 per la prima volta in Toscana Identificazione precoce della malattia Programma di cura prima che si manifestino i sintomi clinici: il decorso clinico della malattia è migliore nei pazienti diagnosticati precocemente Oggi la dx può essere effettuata mediante uno screening neonatale Possibilità di fornire alla coppia di un bambino affetto un’adeguata consulenza genetica

61
Screening neonatale per FC: metodo
Determinazione quantitativa della tripsina immunoreattiva (IRT) su una goccia di sangue prelevata in terza giornata Tale enzima è elevato nelle prime settimane di vita del bambino e poi diminuisce Nei casi sospetti il valore di IRT resta elevato a causa del reflusso di tripsina verso il torrente ematico per ostruzione dei dotti pancreatici Valori elevati di IRT si possono trovare anche in soggetti non affetti da fibrosi cistica. La lattasi può risultare elevata a causa della ridotta funzionalità pancreatica Si tratta di un test poco specifico dato che può dare numerosi falsi positivi e quindi lo si puo’ associare ad un altro test Per ridurre i casi di falsi positivi lo screening affianca il dosaggio della lattasi nel meconio
 su una goccia di sangue prelevata in terza giornata. Tale enzima è elevato nelle prime settimane di vita del bambino e. poi diminuisce. Nei casi sospetti il valore di IRT resta elevato a causa del reflusso di. tripsina verso il torrente ematico per ostruzione dei dotti pancreatici. Valori elevati di IRT si possono trovare anche in soggetti non affetti da fibrosi cistica. La lattasi può risultare elevata a causa della ridotta funzionalità pancreatica. Si tratta di un test poco specifico dato che può dare numerosi falsi positivi e quindi lo si puo’ associare ad un altro test. Per ridurre i casi di falsi positivi lo screening affianca il dosaggio della lattasi nel meconio.")
62
(Enzyme-linked immunoadsorbent assay)
Retesting I soggetti con valori elevati di IRT e lattasi sono sottoposti ad un nuovo test ad un mese di vita, tempo in cui i valori di tali enzimi si normalizzano Elisa (Enzyme-linked immunoadsorbent assay) ELISA è un acronimo che è derivata dall'espressione in inglese Enzyme-Linked ImmunoSorbent Assay (Saggio Immuno-Assorbente legato ad un Enzima). Si tratta di un versatile metodo d'analisi immunologica usato in biochimica per rilevare la presenza di una sostanza usando uno o più anticorpi ad uno dei quali è legato un enzima.
 ELISA è un acronimo che è derivata dall espressione in inglese Enzyme-Linked ImmunoSorbent Assay (Saggio Immuno-Assorbente legato ad un Enzima). Si tratta di un versatile metodo d analisi immunologica usato in biochimica per rilevare la presenza di una sostanza usando uno o più anticorpi ad uno dei quali è legato un enzima.")
63
L’introduzione di questa tecnica ha consentito di effettuare una
VANTAGGI E SVANTAGGI DELLO SCREENING NEOANATALE SULLA FC L’introduzione di questa tecnica ha consentito di effettuare una diagnosi precoce Ampio numero di falsi positivi I soggetti con valori elevati di IRT e lattasi sono sottoposti al test del sudore che può essere effettuato non prima di giorni di vita del bambino

64
Test del sudore Misura la concentrazione del cloro e del sodio escreti nel sudore La diagnosi di FC viene effettata su due risultati positivi ottenuti in due giorni. Caratteristiche cliniche, storia familiare, età del paziente. Il test in assoluto più attendibile per la dx di FC è il test del sudore Le caratteristiche cliniche, la storia familiare e l’età del paziente devono essere tenute in considerazione per l’interpretazione globale dei risultati.

65
TEST DEL SUDORE Valori di riferimento >60 mEq/L test positivo
Sudorazione indotta da iontoforesi pilocarpinica: si stimola farmacologicamente la parte scelta. Misura della quantità di sudore e la concentrazione di cloro in essa presente Valutazione analitica Valori di riferimento >60 mEq/L test positivo 40-60 mEq/L borderline <40 mEq/L test negativo Stimolazione farmacologica del sudore

66
TEST DEL SUDORE Il test del sudore permette di stabilire la diagnosi nella maggioranza dei casi. In alcuni casi il suo significato resta comunque dubbio e l’esame deve essere ripetuto più volte.

67
Fibrosi Cistica:trasmissione
A A A a a A a a Ha una trasmissione autosomica recessiva I portatori hanno il 25% di rischio di avere figli affetti Maschi e femmine sono ugualmente affetti È dovuta all’alterazione di un canale del cloro, denominato CFTR (Cystic Fibrosis Conductance transmembrane regulator) Recessivo: l’allele mutato non si manifesta fenotipicamente se non quando su entrambi gli alleli è presente la mutazione in questione
 Recessivo: l’allele mutato non si manifesta fenotipicamente se non quando su entrambi gli alleli è presente la mutazione in questione.")
68
CF: Genetica Gene CFTR localizzato sul cromosoma 7 (7q31-q32) 27 esoni
>900 mutazioni: elevata variabilità allelica Alcune diffuse in tutto il mondo, altre sono specifiche di alcuni ambiti etnico-geografici Eterogeneità o variabilità allelica: più mutazioni a carico dello stesso locus cromosomico

69
La proteina CFTR Canale per il Cl- delle membrane apicali delle cellule epiteliali Appartiene alla famiglia dei trasportatori glicoproteici di membrana che possiedono siti intracellulari di legame per l’ATP (ABC-family)
")
70
DOVE STA CFTR? Nelle cellule epiteliali del polmone, del tratto digerente, ghiandole del sudore, sistema genitourinario

71
CFTR – IL CANALE DEL CLORO
2 metà omologhe Ogni metà ha 6 domini transmembrana 1 sito di legame per nucleotidi (NBD) collegati da un dominio regolatorio citoplasmatico (R-domain) che contiene i siti di fosforilazione
 collegati da un dominio regolatorio citoplasmatico (R-domain) che contiene i siti di fosforilazione.")
72
CFTR: FUNZIONE Trasporto epiteliale del Cl- L’efficienza del transporto epiteliale di Cl- è determinata dall’attivazione di CFTR che dipende dal suo stato di fosforilazione

73
Dalla mutazione alla malattia
La forma mutata del CFTR impedisce l’uscita del cloro, rendendo viscose le secrezioni Le secrezioni ostruiscono I dotti e alterano la funzionalità di pancreas ed intestino

74
Mutazioni gene CFTR Missenso 41.76 Frameshift 15.76 Splicing 12.71
Nonsenso 9.66 In frame in/del 2.01 Grandi in/del 2.85 Promotore 0.52 Variazioni di sequenza 14.59

75
CFTR defect classification

76
Spettro delle mutazioni CF
Oltre 1000 mutazioni lungo l’intero gene CFTR alterazioni funzionali/strutturali del CFTR F508: 70% alleli FC

77
F508 Presente nel 70% dei pazienti
Delezione di un singolo aminoacido (fenilalanina in pos.508) nell’esone 10 che codifica per la prima parte del NBD-1 di CFTR Produce il misfolding di CFTR nel reticolo endoplasmico (ER) Questa proteina immatura viene degradata dal proteosoma il folding proteico rappesenta Il ripiegamento di proteine o ripiegamento proteico (in inglese protein folding) è il processo di ripiegamento molecolare attraverso il quale le proteine ottengono la loro struttura tridimensionale che implica anche che esse sono attive dal punto di vista funzionale. Una volta mature e attive le proteine vengono rilasciate dal nucleo al reticolo endoplasmatico .
 nell’esone 10 che codifica per la prima parte del NBD-1 di CFTR. Produce il misfolding di CFTR nel reticolo endoplasmico (ER) Questa proteina immatura viene degradata dal proteosoma. il folding proteico rappesenta Il ripiegamento di proteine o ripiegamento proteico (in inglese protein folding) è il processo di ripiegamento molecolare attraverso il quale le proteine ottengono la loro struttura tridimensionale che implica anche che esse sono attive dal punto di vista funzionale. Una volta mature e attive le proteine vengono rilasciate dal nucleo al reticolo endoplasmatico .")
78
CF: F508 Mutazione più comune Etnia F508 frequenza
Afro-Americani 30% Ebrei Ashkenazi 37% Spagnoli/Italiani 50% Canadesi 70% Caucasici nord americani 76%

79
Epidemiologia delle mutazioni CFTR in Italia

80
CF: Indicazioni al test genetico
Individui affetti Carrier test Partners di individui con CF Diagnosi prenatale

81
SCREENING di PRIMO LIVELLO
Ricerca delle mutazioni più frequenti causative della fibrosi cistica mediante l’utilizzo di Kit diagnostici che sfruttano il principio della ibridazione allele-specifica (ASO).
.")
82
SCREENING di PRIMO LIVELLO:
tipo di campione da cui partire Prelievo sangue venoso, o della mucosa buccale, villi coriali, liquido amniotico Estrazione del DNA Multiplex PCR delle regioni geniche Ibridazione DNA con sonde allele specifiche RIVELAZIONE

83
Metodica Reverse dot blot
SCREENING di PRIMO LIVELLO: Metodica Reverse dot blot Le regioni del gene CFTR, dove sono localizzate le mutazioni da analizzare, sono amplificate simultaneamente mediante l’impiego di specifiche coppie di oligonucleotidi. Dopo la multilex PCR, l’amplificato viene denaturato e posto su una striscia di nitrocellulosa su cui sono presenti le sonde allele specifiche da testare. Tramite una reazione colorimetrica (biotina-streptavidina) si forma un precipitato scuro che permette di visualizzare, per ogni tratto di DNA, una o due bande colorate che consentiranno di stabilire la presenza o meno della mutazione testata e lo stato di omozigosi o eterozigosi
 si forma un precipitato scuro che permette di visualizzare, per ogni tratto di DNA, una o due bande colorate che consentiranno di stabilire la presenza o meno della mutazione testata e lo stato di omozigosi o eterozigosi.")
84
RDB - REVERSE DOT BLOT:ibridazione inversa degli acidi nucleici
Le sonde allele specifiche vengono fatte aderire stabilmente a una membrana di nylon. Dopo l’ibridazione tra le sonde e il DNA amplificato, nel quale è incorporato dUTP biotinilato, si procede alla visualizzazione colorimetrica con una reazionedi tipo biotina-streptavidina-fosfatasi alcalina.

85
Detection omozigote MM eterozigote NM omozigote NN

86
Kit Diagnostici

87
RDB MULTIPLO analisi di 36 mutazioni della Fibrosi Cistica (gene CFTR)
")
88
Le mutazioni identificate devono essere successivamente
confermate mediante sequenziamento diretto

89
Indagine prenatale di Fibrosi Cistica
(analisi diretta)
")
90
ANALISI II LIVELLO PCR dei 27 esoni DHPLC 12 esoni a 57°C 15 esoni
Ex 1,3,4,8,13,14a,15, 16,18,19,20,22,23 Sequenziamento Ex 2,5,6a,6b,7,9,10,11, 12,14b,17a,17b,21,24 Analisi di II livello: scanning di tutti gli esoni e delle regioni limitrofe, riconoscimento di variazioni di sequenza, sequenziamento della specifica regione del gene. Le tecniche più utilizzate sono: Denaturing Gradient Gel Electrophoresis e Denaturing High Performance Liquid Cromatography. I test di II livello permettono un tasso di individuazione (detection rate) migliore, ma il significato fenotipico del risultato molecolare può essere di difficile interpretazione. Per molte mutazioni CFTR le consegenze funzionali sono sconosciute si potrebbe trattare di polimorfismi. Inoltre questi saggi screenano per mutazioni presenti nelle regioni esoniche e nelle giunzioni esone/introne del gene CFTR. Mutazioni localizzate nel promotore o nelle regioni introniche sfuggono a questo tipo di analisi
 migliore, ma il significato fenotipico del risultato molecolare può essere di difficile interpretazione. Per molte mutazioni CFTR le consegenze funzionali sono sconosciute si potrebbe trattare di polimorfismi. Inoltre questi saggi screenano per mutazioni presenti nelle regioni esoniche e nelle giunzioni esone/introne del gene CFTR. Mutazioni localizzate nel promotore o nelle regioni introniche sfuggono a questo tipo di analisi.")
91
Analisi di III livello: Ricerca di macrodelezioni CFTR
Nel 10-15% dei soggetti affetti non si riesce a identificare la o le mutazioni responsabili della malattia. Utilizzo di tecniche in grado di identifcare i riarrangiamenti genomici che coinvolgono delezioni di più esoni in uno stesso gene

92
Ricerca di macrodelezioni CFTR
MLPA=Multiplex Ligation-dependent Probe Amplification Permette di stabilire il numero di copie di ciascun esone del gene CFTR e quindi di caratterizzare delezioni e duplicazioni I prodotti di amplificazione vengono separati su elettroforesi capillare Il confronto tra il pattern di amplificazione del campione di interesse e di controllo indica quale/i esone/i mostrano un numero aberrante di copie Il DNA di ogni paziente è stato analizzato mediante un kit commerciale basato su PCR quantitativa seguita da elettroforesi capillare (MLPA SALSA kit, MRC-Holland, The Netherlands) (3). La tecnica MLPA (multiplex ligation-dependent probe assay) permette di analizzare i 27 esoni del gene CFTR mediante l’amplificazione sequenza specifica e simultanea effettuata con un set di 44 probe sintetici.
 (3). La tecnica MLPA (multiplex ligation-dependent probe assay) permette di analizzare i 27 esoni del gene CFTR mediante. l’amplificazione sequenza specifica e simultanea effettuata con un. set di 44 probe sintetici.")
93
Ibridazione & Ligazione
MLPA probemix è costituito da 43 sonde disegnate per ciascuno dei 27 esoni del gene e per geni di controllo Viene aggiunto al DNA genomico denaturato Ciascuna sonda ibridizza due sequenze adiacenti

94
Ibridazione & Ligazione
4. La ligazione delle sonde avviene tramite una ligasi termostabile

95
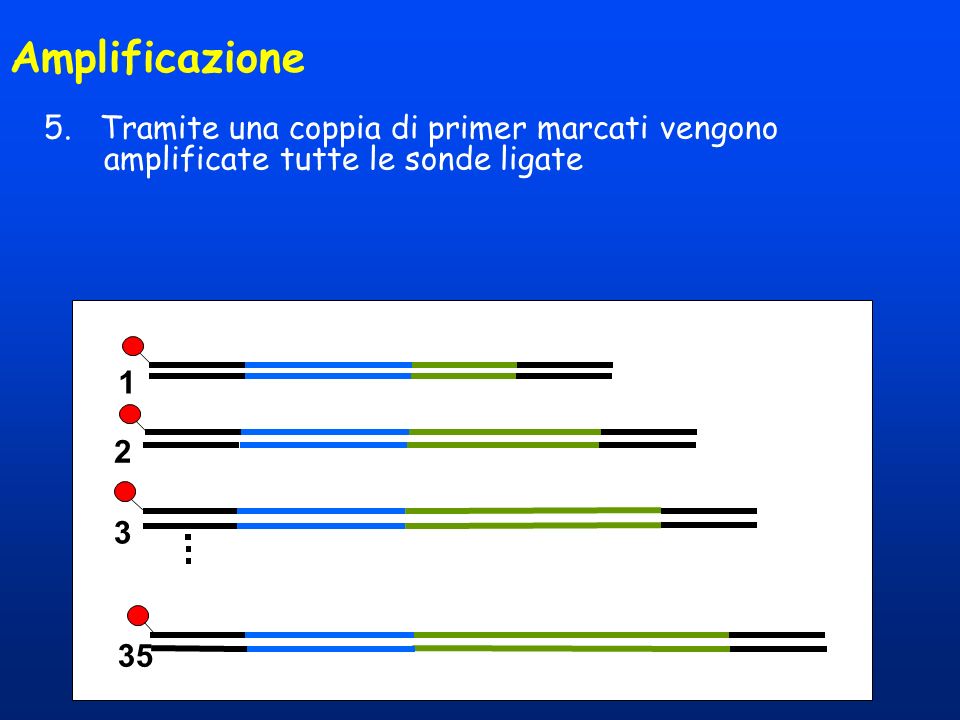
Amplificazione 5. Tramite una coppia di primer marcati vengono amplificate tutte le sonde ligate 1 2 3 35
96
Elettroforesi capillare
CFTR delex2,3

97
LA DIAGNOSI PRENATALE nella FC
Nelle coppie a rischio Identificazione della mutazione nei genitori Ricerca della mutazioni parentali nel feto

98
Analisi Diretta I Livello: Reverse Dot Blot
- Analisi delle mutazioni più frequenti II Livello: - DHPLC - SEQUENZIAMENTO III Livello: MLPA

99
Analisi Indiretta Ripetizioni dei polimorfismi più frequenti
Utilizzo della segregazione dei polimorfismi IVS8GT e IV17bCA etc che sono considerati come i più frequenti. Si vanno a valutare il numero di ripetizioni di questi polimorfismi Ripetizioni dei polimorfismi più frequenti

100
Cura Anche se attualmente nessuna cura è in grado di guarire completamente la Fibrosi Cistica, numerose terapie permettono di contrastare l’evoluzione della malattia, controllando le infezioni polmonari, fornendo un’alimentazione adeguata e prevenendo l’ ostruzione intestinale. Fisioterapia e riabilitazione respiratoria: per rimuovere dalle vie respiratorie il muco che le ostruisce e che favorisce le infezioni. Aerosolterapia: per fluidificare il muco o somministrare antibiotici per via aerea per controllare le infezioni respiratorie croniche. Antibioticoterapia: per bocca o per via endovenosa, a cicli o per periodi molto prolungati, anche in continuazione, per eliminare o contenere la carica e l'aggressività dei batteri. Il problema associato alla terapia genica è sempre quello legato alla possib. Di veicolare la copia corretta gel gene senza scatenare reazioni di rigetto da parte dell’organismo Nutrizione: alimentazione sostenuta, ipercalorica, ricca di grassi associata a somministrazione di enzimi pancreatici ad ogni pasto, in sostituzione di quelli che il pancreas non produce, e integrata da vitamine liposolubili.

101
Terapia genica La terapia genica mira nell’uso della forma normale del gene allo scopo di correggere il gene mutato causativo della malattia. Il goal sarebbe quello di sostituire il gene mutato nelle cellule del polmone per curare la malattia o per diminuirne la progressione. Vettori per trasferire la proteina: virus adenoassociati, liposomi, cellule staminali. Correzioni della funzionalità della proteina legata al suo cattivo ripiegamento. Aumento del numero dei canali del cloro funzionanti

Presentazioni simili














>")




