Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

2
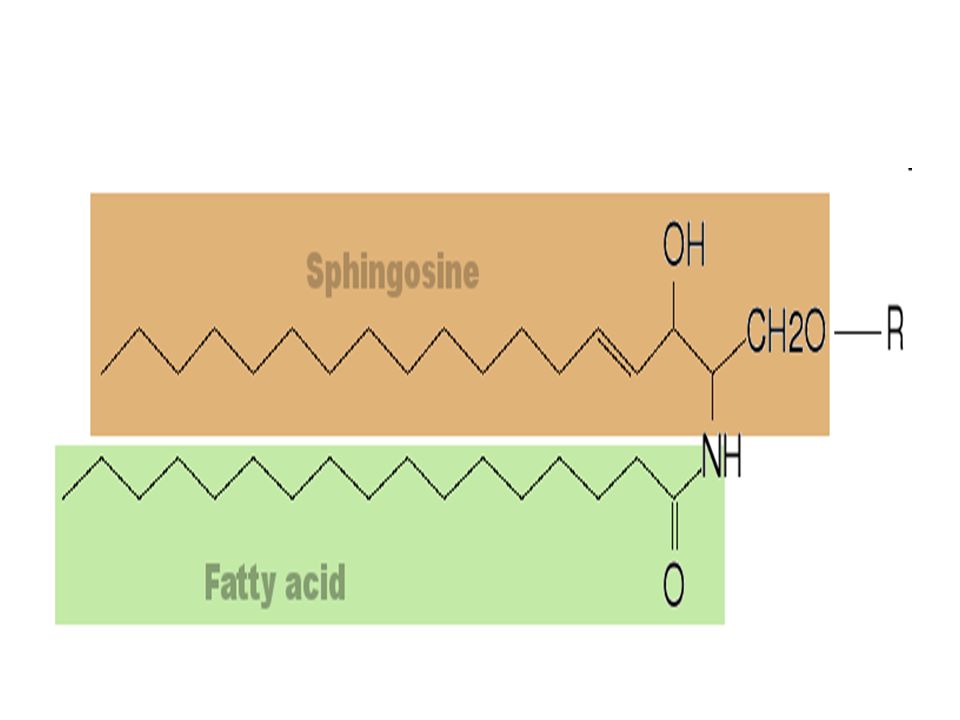
Le ceramìdi sono una famiglia di molecole lipidiche
Le ceramìdi sono una famiglia di molecole lipidiche. Una ceramide è composto di sfingosina e acido grasso. Ceramidi si trovano in alta concentrazione nelle membrane cellulari. Sono una delle componenti lipidiche fonte di sfingomielina: uno dei principali lipidi dello doppio strato lipidico delle membrane cellulari,

5
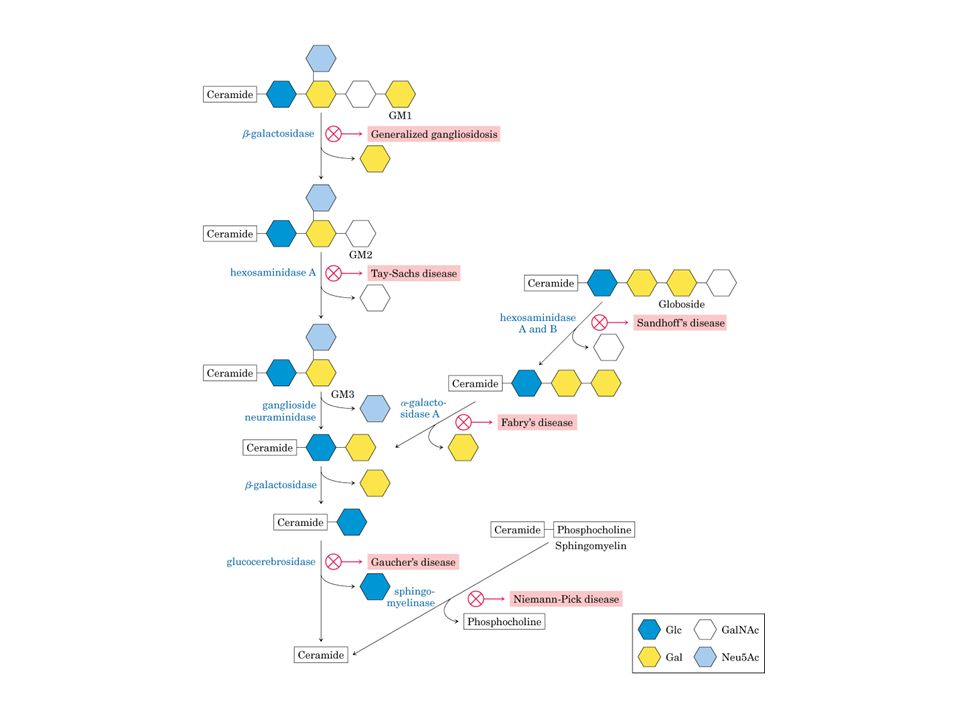
Le gangliosidosi GM2 Sono malattie neurodegenerative prodotte da una diminuita capacità di metabolizzare i gangliosidi GM2 che, in normali condizioni, sono idrolizzati dall’isoenzima A della beta-N-acetil-esosaminidasi (EC ). L’enzima spesso indicato con la sigla Hex è lisosomiale e partecipa alla degradazione delle glicoproteine, dei glicolipidi, dei glicosaminoglicani poiché la sua attività catalitica si esplica sull’idrolisi del legame beta-glicosidico della N-acetilglucosammina e della N-acetil galattosammina
. L’enzima spesso indicato con la sigla Hex è lisosomiale e partecipa alla degradazione delle glicoproteine, dei glicolipidi, dei glicosaminoglicani poiché la sua attività catalitica si esplica sull’idrolisi del legame beta-glicosidico della N-acetilglucosammina e della N-acetil galattosammina.")
6
esosaminidasi LE ESOSAMINIDASI
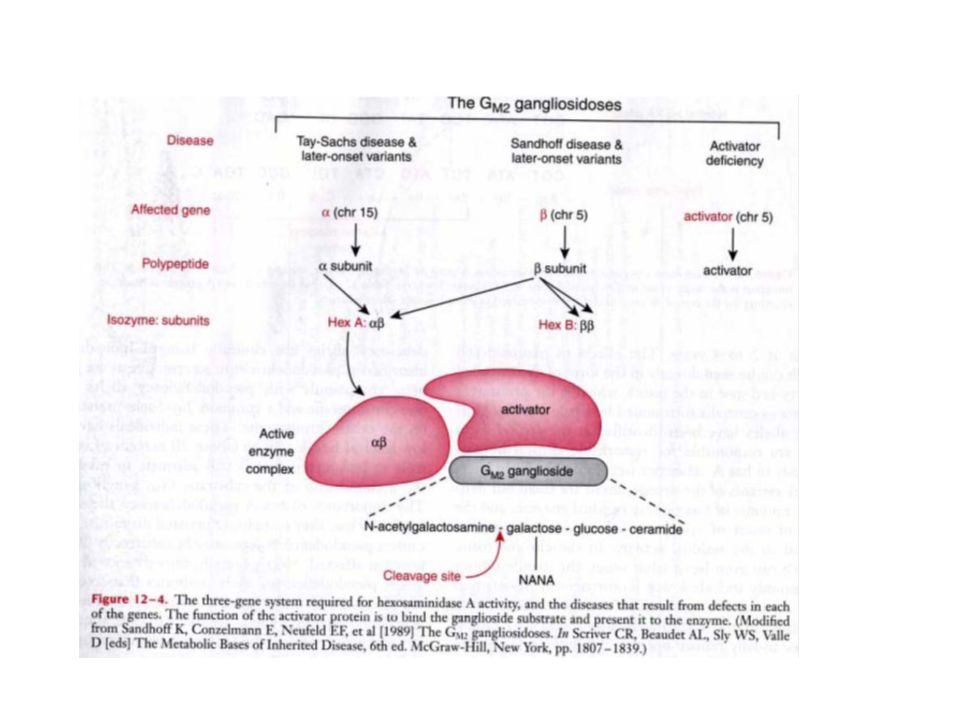
L’esosaminidasi A esosaminidasi è un enzima lisosomiale che, con il cofattore GM2 activator protein , catalizza la degradazione , del ganglioside GM2 e di altre molecole che hanno N- acetil esosamine terminali. La esosaminidasi A è costituita da due subunit subunità, alfa e beta, codificate da geni distinti , HEX-A ed HEX-B). Le subunità alfa e beta sono membri della famiglia delle glicosil idrolasi. Mutazioni nei geni della subunit subunità alfa o beta determinano un accumulo di ganglioside GM2 nei neuroni e di conseguenza alterazioni neurodegenerative (le gangliosidosi GM2). Mutazioni a carico del gene per la subunità alfa determinano la malattia di Tay-Sachs (GM2-gangliosidosi di tipo I). L’esosaminidasi B esosaminidasi è un omodimero di subunità beta.
. Le subunità alfa e beta sono membri. della famiglia delle glicosil idrolasi. Mutazioni nei geni. della subunit subunità alfa o beta determinano un accumulo di. ganglioside GM2 nei neuroni e di conseguenza alterazioni. neurodegenerative (le gangliosidosi GM2). Mutazioni a. carico del gene per la subunità alfa determinano la. malattia di Tay-Sachs (GM2-gangliosidosi di tipo I). L’esosaminidasi B esosaminidasi è un omodimero di subunità beta.")
7
L’enzima esosaminidasi (Hex)
Hex ha una struttura dimerica composta da due catene non identiche (α e β). Il grado di omologia è di circa il 60%. Esiste in due isoenzimi principali: HexA che è un eterodimero αβ e HexB che è un omodimero ββ. Esiste anche un terzo isoenzima poco stabile αα, che raggiunge livelli del 5% solo quando la subunità β non viene sintetizzata come succede nella malattia di Sandhoff.
. Il grado di omologia è di circa il 60%. Esiste in due isoenzimi principali: HexA che è un eterodimero αβ e HexB che è un omodimero ββ. Esiste anche un terzo isoenzima poco stabile αα, che raggiunge livelli del 5% solo quando la subunità β non viene sintetizzata come succede nella malattia di Sandhoff.")
9
L’enzima esosaminidasi (Hex)
Il sito attivo della subunità α idrolizza preferibilmente substrati provvisti di carica negativa come il GM2 anche se può idrolizzare anche substrati neutri. Al contrario l’attività catalitica della subunità β si esplica solo su substrati neutri, cioè l’Hex B non agisce sui gangliosidi GM2.

10
L’enzima esosaminidasi (Hex)
Come per molti altri enzimi lisosomiali la relativa semplicità della reazione catalizzata dall’ HEX contrasta con il complesso processo di traslocazione del suo precursore ad alto peso molecolare dal reticolo endoplasmatico ai ribosomi dove viene immagazzinato nella sua forma a più basso peso molecolare. Queste trasformazioni delle catene polipeptidiche non sono prerequisiti per la piena attività catalitica che, infatti, è posseduta anche dei precursori, ma le subunità DEVONO dimerizzare per poter possedere l’attività catalitica.

11
L’enzima esosaminidasi (Hex)
La dimerizzazione delle catene è un prerequisito per l’attività enzimatica. La dimerizzazione ββ è più veloce di quella αβ. Così può sucecdere che all’interno dei lisosomi si trovino catene α non assemblate. Le catene assemblate vengono trasferite nel Golgi dove vengono aggiunte di mannosio 6 fosfato che è un marker di riconoscimento per maturare la proteina finale.

12
DEFICIT ENZIMATICO Le Gangliosidosi GM2 sono disordini lisosomiali che si trasemttono in modo autosomico recessivo e sono caratterizzate dalla incapacità di idrolizzare il ganglioside GM2 che viene accumulato in particolare nelle cellule neuronali. La deficienza di HexA (αβ) e di HexB(ββ) prodotta da differenti mutazioni sul gene che codifica per le subunità β causa la gangliosidosi di tipo 0 detta anche malattia di Sandhoff). Una deficienza di HexA prodotta da mutazioni sul gene che codifica per la subunità α, causa la malattia di Tay-Sachs che coinvolge due tipi enzimologici diversi: La variante B caratterizzata dalla perdita dell’HexA La variante B1caratterizzata dall’incapacità di HexA di idrolizzare substrati negativi ma di poter agire su quelli neutri. In realtà l’attività enzimatica ha bisogno di un attivatore proteico (GM2 activator protein) la cui mutazione provoca gangliosidosi GM2 che però è molto più difficile da diagnosticare
 e di HexB(ββ) prodotta da differenti mutazioni sul gene che codifica per le subunità β causa la gangliosidosi di tipo 0 detta anche malattia di Sandhoff). Una deficienza di HexA prodotta da mutazioni sul gene che codifica per la subunità α, causa la malattia di Tay-Sachs che coinvolge due tipi enzimologici diversi: La variante B caratterizzata dalla perdita dell’HexA. La variante B1caratterizzata dall’incapacità di HexA di idrolizzare substrati negativi ma di poter agire su quelli neutri. In realtà l’attività enzimatica ha bisogno di un attivatore proteico (GM2 activator protein) la cui mutazione provoca gangliosidosi GM2 che però è molto più difficile da diagnosticare.")
13
FENOTIPI CLINICI Il fenotipo clinico delle gangliosidosi GM2 varia considerevolmente da una forma infantile fino a quella adulta. In generale tutte le gangliosidosi hanno gli stessi sintomi: alterazione del linguaggio, distonia, convulsioni, eccetera.

14
*606869*606869HEXOSAMINIDASE A; HEXAHEXOSAMINIDASE HEXAGene map locus Gene 15q2315q23--q24q24
IL GENE IL Il gene HEXA codifica la subunità αdelldell’’esosaminidasi A (EC EC ), un enzima, lisosomiale coinvolto nella degradazione dei gangliosidi. LA MALATTIA La malattia di Tay--Sachs è un’alterazione neurodegenerativa progressiva autosomica recessiva che, nella forma infantile classica, è letale entro il 2°o 3°anno d’età . E’caratterizzata dalla comparsa, nella prima infanzia, di un ritardo nello sviluppo mentale, seguito da paralisi, demenza e cecità, e dalla morte prematura. Un segno caratteristico all’’esame del fondo dell’occhio è la presenza di unla un’area bianco--grigiastra attorno alla fovea centrale, causata da cellule gangliari cariche di lipidi, con una macchia rosso ciliegia”. La verifica isto--patologica conferma la presenza di neuroni di forma tipicamente arrotondati al livello del sistema nervoso a centrale. E’presente molto precocemente un’ esagerata risposta ai rumori ('startle reaction') che permette di riconoscere la malattia. TAYTAY--SACHS DISEASE
, un enzima, lisosomiale coinvolto nella degradazione dei gangliosidi. LA MALATTIA La malattia di Tay--Sachs è un’alterazione neurodegenerativa progressiva autosomica recessiva che, nella forma infantile classica, è letale entro il 2°o 3°anno d’età . E’caratterizzata dalla comparsa, nella prima infanzia, di un ritardo nello sviluppo mentale, seguito da paralisi, demenza e cecità, e dalla morte prematura. Un segno caratteristico all’’esame del fondo dell’occhio è la presenza di unla un’area bianco--grigiastra attorno alla fovea centrale, causata da cellule gangliari cariche di lipidi, con una macchia rosso ciliegia . La verifica isto--patologica conferma la presenza di neuroni di forma tipicamente arrotondati al livello del sistema nervoso a centrale. E’presente molto precocemente un’ esagerata risposta ai rumori ( startle reaction ) che permette di riconoscere la malattia. TAYTAY--SACHS DISEASE.")
15
LA MALATTIA di Sandhoff La è un’alterazione neurodegenerativa progressiva caratterizzata dall’accumulo di gangliosidi GM2, particolarmente nei neuroni. Il quadro clinico e patologico è molto simile a quello della malattia di Tay--Sachs Estrema debolezza ne. primi 6 mesi di vita. Startle reaction, cecità precoce, progressivo deterioramento mentale e fisico, macchie rosso ciliegia e macrocefalia, HEXBHEXOSAMINIDASE HEXBGene map locus Gene 5q135q13 IL GENE IL Il gene HEXB codifica la subunità β dell’esosaminidasi A (EC EC ), un enzima ), lisosomiale coinvolto nella degradazione dei gangliosidi.. SANDHOFF DISEASEDISEASE
, un enzima ), lisosomiale coinvolto nella degradazione dei gangliosidi.. SANDHOFF DISEASEDISEASE.")
16
Caratterizzazione biochimica
L’Hexb è termostabile l’Hex A no Quindi si può determinare prima l’attività enzimatica totale, poi si inattiva al calore e si rimisura l’attività con un substrato fluorogenico:4-Methylumbelliferyl-N-acetyl-β-D-glucosaminide

17
Caratterizzazione biochimica
A volte il dosaggio su-descritto può dare una sovrastima dell’HexA e il dosaggio immunologico non è adatto perché da un punto di vista immunologico l’enzima inattivo è indistinguibile da quello attivo La diagnosi della forma B1 si a con un substrato fluorigenico negativo methyl lumbelliferyl glucosaminidine 6 sulphate che comunque è molto costoso specie per poter fare indagini su popolazioni per rilevare l’eterozigosi

18
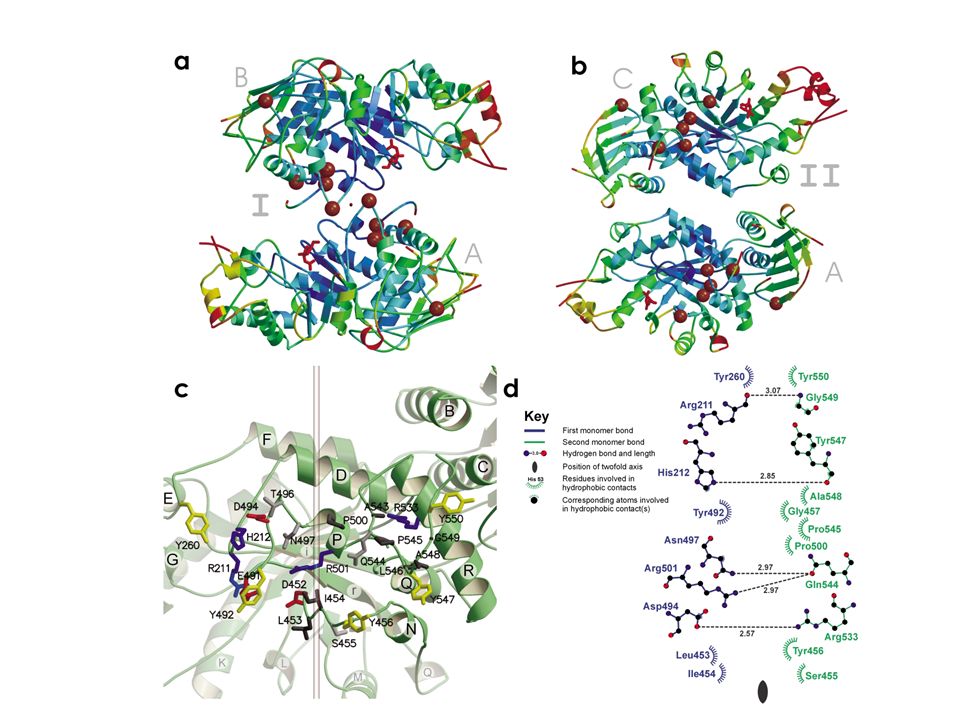
Struttura cristallografica
E’ stata determinata la struttura cristallografica dell’HexB ricombinante in cellule di insetto.

19
Figura a: i cerchi sono le alfa eliche, i triangoli i beta foglietti, i numeri sono le posizioni nella sequenza, gli esagoni i siti di glicosilazione e in rosso i ponti disolfuro. I cerchietti gialli sono i residui del sito attivo.

20
Sito catalitico: Glu355 .

21
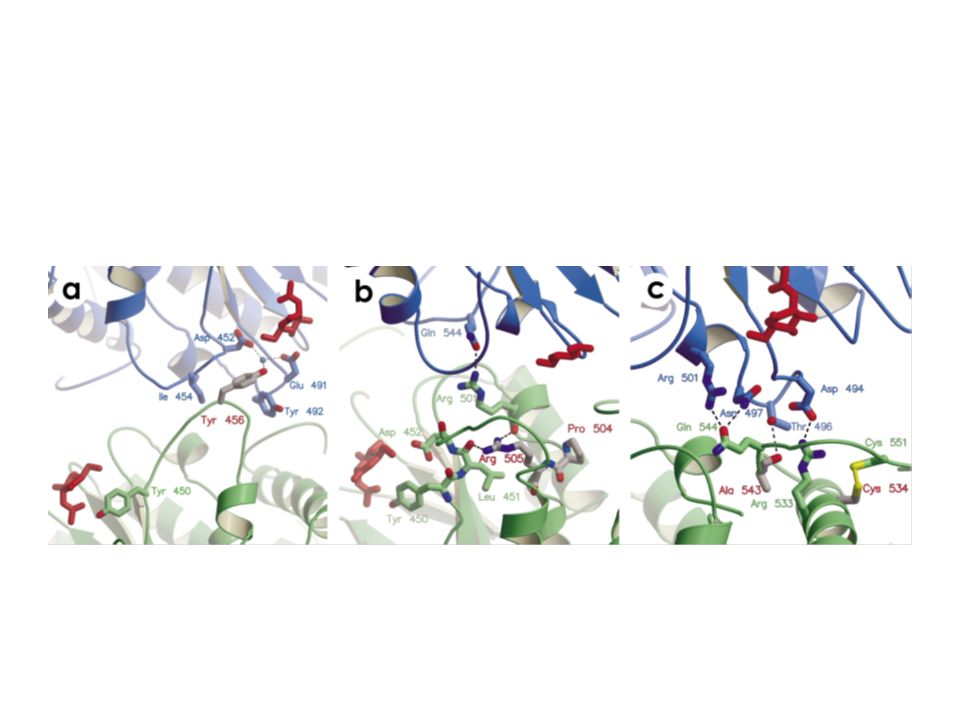
L’interfaccia del dimero
E’ composta da le regioni C terminali dell’αβ barrell. I loops sono determinati dall’elica D E e F. Sono presenti 5 tirosine e legami polari tra leucine (453 e 546) e prolina (545)
 e prolina (545)")
23
mutazioni C185T ch porta alla sostituzione della Ser62 in Leu: si è ritrovata nella malattia di Sandhoff infantile La Serina è un residuo che favorisce la formazione di un beta-turn all’Nterminale del beta-strand a. La leucina lo sfavorisce La serina è importante per il fold della proteina

24
mutazioni C1214T nell’esone 11 porta alla produzione di un mRNA instabile e con uno splicing errato cosicchè i livelli di HexB risultano molto diminuiti Pro417Leu e Cys309Tyr sono state osservate assieme in alcune pazienti. La cisteina fa i ponti disolfuro che stabilizzano la struttura

25
mutazioni Arg505Gln è alla base della forma adulta
L’enzima mutato è instabile al calore L’Arg 505 è localizzata all’N terminale dell’elicaQ e forma un labile legame idrogeno con la Leu451 (ossigeno peptidico). La Leu451 ha una vicinanza diretta con la Tyr450 e l’Asp452 che giocano un ruolo importante nel legame col substrato.
. La Leu451 ha una vicinanza diretta con la Tyr450 e l’Asp452 che giocano un ruolo importante nel legame col substrato.")
27
TAY-SACHS

28
TAY-SACHS

29
TAY-SACHS Studi recenti hanno dimostrato una certa eterogeneità all’interno delle deficienze della HexA con diversi tipi di mutazione. Oltre a quella descritt prima in popolazioni Francesi-canadesi si ha una prevalenza della delezione del primo esone o delle sequenze vicine così da produrre l’assenza dell’mRNA corrispondente alla subunità α e quindi la malattia di Tay-Sachs infantile.

30
TAY-SACHS Altre mutazioni non associate con popolazioni sono: la sostituzione della lisina482 con l’acido glutammico e la delezione della citosina in posizione 504 con il risultato di una prematura terminazione. In ambedue i casi la proteina risulta difettosa per il suo trasporto intracellulare dal reticolo endoplasmatico al Golgi. E queste sono varianti B

31
TAY-SACHS Tra le varianti B1 si ricorda la sostituzione dell’istidina 168 con l’arginina che abolisce l’attività catalitica nei confronti dei GM2

32
Effective gene therapy in an authentic model of Tay-Sachs-related diseases
Mice lacking Hex A and B activity as a result of targeted disruption of the hex β subunit gene (Sandhoff strain) provide an authentic model of acute human Tay-Sachs disease and are invaluable for evaluation of innovative therapies. Indistinguishable from wild-type and heterozygous littermates at birth, homozygous hexb −/− mice grow normally to early adulthood. Neurological impairment is apparent at 3 months with head tremor, followed by ataxia, bradykinesia, impaired power, and balance. By 4.5 months, hind-limb movement is lost; Sandhoff mice die before the age of 5 months
 provide an authentic model of acute human Tay-Sachs disease and are invaluable for evaluation of innovative therapies. Indistinguishable from wild-type and heterozygous littermates at birth, homozygous hexb −/− mice grow normally to early adulthood. Neurological impairment is apparent at 3 months with head tremor, followed by ataxia, bradykinesia, impaired power, and balance. By 4.5 months, hind-limb movement is lost; Sandhoff mice die before the age of 5 months.")
33
Effective gene therapy in an authentic model of Tay-Sachs-related diseases
The molecular pathogenesis of brain injury associated with GM2 storage in humans and animals is ill understood. Disruption of intracellular structures by the expanded lysosomal compartment, enhanced excitatory neurotransmitter activity with abnormal neurite outgrowth, and the accumulation of cytotoxic metabolites may all contribute. Impaired generation of isoglobotrihexosylceramide in Sandhoff mice causes depletion of natural killer T (NKT) cells, a distinct lineage of T cells, indicating that presentation of lysosomal ligands is defective, thereby altering NKT cell responses to infection, malignancy, and immune recognition.
 cells, a distinct lineage of T cells, indicating that presentation of lysosomal ligands is defective, thereby altering NKT cell responses to infection, malignancy, and immune recognition.")
34
Effective gene therapy in an authentic model of Tay-Sachs-related diseases
With widespread inflammation and storage of GM2 and other gangliosides, the Tay-Sachs-related diseases present formidable challenges for treatment. Although allogeneic bone marrow transplantation has little benefit in human GM2 gangliosidosis, it partially repopulates the nervous system with healthy donor macrophages and ameliorates disease in the Sandhoff mouse . The use of inhibitors of glycosphingolipid synthesis to balance formation with the impaired catabolism also delays the onset of neurological signs in Sandhoff mice. However, in the absence of appreciable hexosaminidase activity in this knockout strain or in the acute human GM2 gangliosidoses, monotherapy with substrate-reducing agents will not arrest the disease.

35
Effective gene therapy in an authentic model of Tay-Sachs-related diseases
M. Begoña Cachón-González et al report the use of gene therapy delivered directly to the brain for murine GM2 gangliosidosis. Adult Sandhoff mice were stereotaxically inoculated with recombinant adeno-associated viral (rAAV) vectors encoding complementing human β-hexosaminidase α and β subunits (rAAVα and rAAVβ). Widespread and sustained expression of hexosaminidase activity in the brain and spinal cord reduced pathological storage and inflammation. When onset of neurological signs was delayed markedly and the pattern of disability was attenuated, survival was greatly prolonged.
 vectors encoding complementing human β-hexosaminidase α and β subunits (rAAVα and rAAVβ). Widespread and sustained expression of hexosaminidase activity in the brain and spinal cord reduced pathological storage and inflammation. When onset of neurological signs was delayed markedly and the pattern of disability was attenuated, survival was greatly prolonged.")
Presentazioni simili





>")














