Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Consideriamo un cristallo di Ag (fcc; a=4.09Å), descritto da un qualche potenziale semiempirico (negli esempi sarà EAM, ma non importa). Vogliamo studiarne delle quantità di bulk: quanto grande dobbiamo prendere la cella di simulazione?

2
4 atomi (c.conv.) 32 atomi 108 atomi 256 atomi 864 atomi 2048 atomi 4000 atomi
 32 atomi 108 atomi 256 atomi 864 atomi 2048 atomi 4000 atomi")
3
Atomi L(Å)
")
4
Atomi A(Å 2 )
")
5
Atomi V(Å 3 )
")
6
Å

7
Spostamenti da posizioni di bulk dopo minimizzazione energia (4 atomi; non è nemmeno un fcc..)
")
8
Spostamenti da posizioni di bulk dopo minimizzazione energia (4 atomi)
")
9
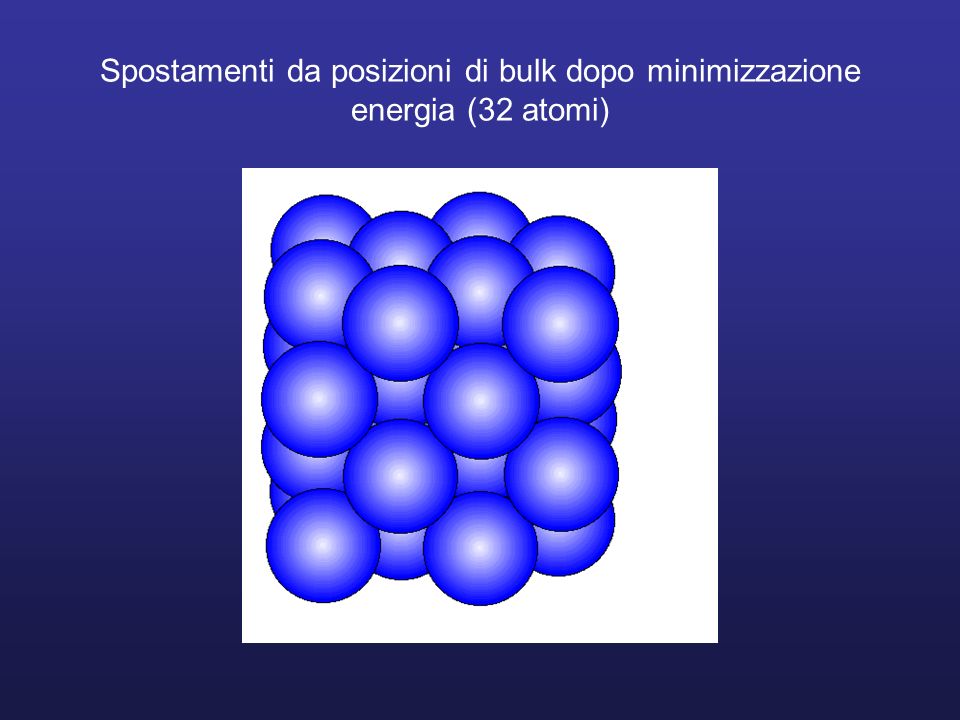
Spostamenti da posizioni di bulk dopo minimizzazione energia (32 atomi)
")
11
Spostamenti da posizioni di bulk dopo minimizzazione energia (4000 atomi)
")
13
Chiaramente, ho sempre degli effetti di superficie: posso sperare di individuare una “zona di bulk” all’interno? Atomi A/V

14
32 atoms r(Å) bonds % atoms in perfect positions: 0
 bonds % atoms in perfect positions: 0")
15
108 atoms r(Å) bonds % atoms in perfect positions: 0
 bonds % atoms in perfect positions: 0")
16
256 atoms r(Å) bonds % atoms in perfect positions: 3
 bonds % atoms in perfect positions: 3")
17
864 atoms r(Å) bonds % atoms in perfect positions: 7
 bonds % atoms in perfect positions: 7")
18
2048 atoms r(Å) bonds % atoms in perfect positions: 11
 bonds % atoms in perfect positions: 11")
19
4000 atoms r(Å) bonds % atoms in perfect positions: 15
 bonds % atoms in perfect positions: 15")
20
Esiste un metodo alternativo a costruire sistemi sempre più grandi per cercare di eliminare gli effetti di superficie (a parte “trucchi” come la saturazione dei legami nei semiconduttori)?
")
21
YES! LET’s GO PBC

22
Periodic boundary conditions ? ?

25
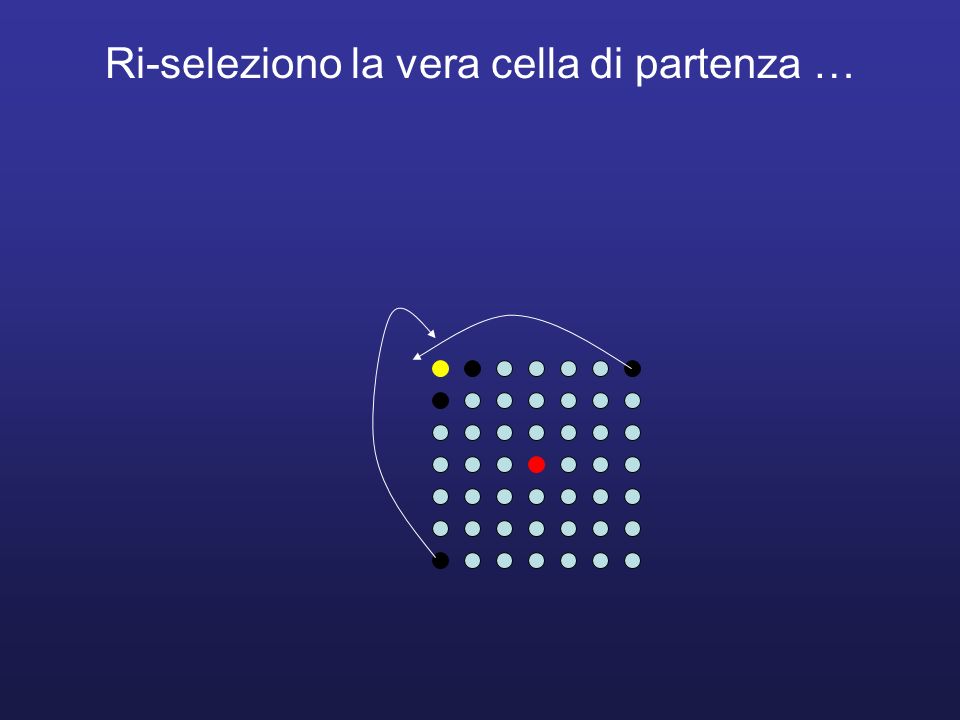
Ri-seleziono la vera cella di partenza …

27
a d=a d=2a L=7a d=3a d=4a; dimm=3a d=5a; dimm=2a d=6a; dimm=a

28
Ci siamo concentrati sui primi vicini. In realtà se uso un potenziale “privo di cutoff” come Lennard-Jones, se ripeto la cella ogni atomo sarà influenzato dalle proprie immagini. Se non studio situazioni in cui tutti gli atomi si comportano in maniera identica (proprietà statiche di un cristallo perfetto) NON VA BENE!
 NON VA BENE!.")
29
a d=a d=2a L=7a d=3a d=2a d=a Minimum image convention: definisco distanza d tra l’atomo i e l’atomo j della cella originale la distanza tra i e l’immagine di j che minimizza d. Nei conti, considero solo l’interazione tra i e tale immagine. Di fatto questo modo di procedere un po’ arbitrario diventa più chiaro se forzo un cutoff preciso per il potenziale. Per esempio, se il cutoff è pari alla distanza tra nn, risulta chiaro con chi può interagire l’atomo giallo Implementazione in fortran: è banale!!!!!!!

30
L=5a 1 2 3 4 5 x(2)-x(1)=a x(3)-x(1)=2a x(4)-x(1)=3a x(5)-x(1)=4a Voglio calcolare la separazione (con segno) tra un atomo e l’atomo 1 (giallo). In assenza di PBC è banale. Con le PBC ….

31
L=5a 1 2 3 4 5 L=5a 1’ 2’ 3’ 4’ 5’ PBC + minimum image convention x(2)-x(1)=a x(2’)-x(1)=-4a -> x(2,MI)-x(1)=a x(3)-x(1)=2a x(3’)-x(1)=-3a -> x(3,MI)-x(1)=2a x(4)-x(1)=3a x(4’)-x(1)=-2a -> x(4,MI)-x(1)=-2a x(5)-x(1)=4a x(5’)-x(1)=-a -> x(5,MI)-x(1)=-a Occhio che cambia tutto. Con un cutoff a primi vicini, e senza PBC, l’atomo 1 interagisce solo con 2. Con le PBC interagisce anche con 5 (tramite la sua immagine). Se non avessi cutoff nel potenziale, l’utilizzo della minimum image convention di fatto ne introduce uno, pari alla metà della cella. Ma è abrupto. Molto meglio usare una cella grande e imporre un vero cutoff (vedi dopo).
. Se non avessi cutoff nel potenziale, l’utilizzo della minimum image convention di fatto ne introduce uno, pari alla metà della cella. Ma è abrupto. Molto meglio usare una cella grande e imporre un vero cutoff (vedi dopo)..")
32
L=5a 1 2 3 4 5 L=5a 1’ 2’ 3’ 4’ 5’ PBC + minimum image convention x(2)-x(1)=a x(2’)-x(1)=-4a -> x(2,MI)-x(1)=a x(3)-x(1)=2a x(3’)-x(1)=-3a -> x(3,MI)-x(1)=2a x(4)-x(1)=3a x(4’)-x(1)=-2a -> x(4,MI)-x(1)=-2a x(5)-x(1)=4a x(5’)-x(1)=-a -> x(5,MI)-x(1)=-a Considero la funzione anint, che approssima con l’intero più vicino. Suppongo di calcolare un certo dx, poi calcolo anint (dx/L). anint(dx/cella)=0 se |dx|<L/2, =1 altrimenti. Bene, se allora prima calcolo dx e poi lo sostituisco con dx=dx-L*anint(dx/L) sono a posto!!!!! Es. dx=3a; anint 3/5=1; dx=3a-5a=-2a Es. dx=2a; anint 2/5=0; dx=2a
. anint(dx/cella)=0 se |dx|<L/2, =1 altrimenti. Bene, se allora prima calcolo dx e poi lo sostituisco con dx=dx-L*anint(dx/L) sono a posto!!!!. Es. dx=3a; anint 3/5=1; dx=3a-5a=-2a Es. dx=2a; anint 2/5=0; dx=2a.")
33
L=5a 1 2 3 4 5 L=5a 1’ 2’ 3’ 4’ 5’ Considero la funzione anint, che approssima con l’intero più vicino. Suppongo di calcolare un certo dx, poi calcolo anint (dx/L). anint(dx/cella)=0 se |dx|<L/2, =1 altrimenti. Bene, se allora prima calcolo dx e poi lo sostituisco con dx=dx-L*anint(dx/L) sono a posto!!!!! Es. dx=3a; anint 3/5=1; dx=3a-5a=-2a Es. dx=2a; anint 2/5=0; dx=2a
. anint(dx/cella)=0 se |dx|<L/2, =1 altrimenti. Bene, se allora prima calcolo dx e poi lo sostituisco con dx=dx-L*anint(dx/L) sono a posto!!!!. Es. dx=3a; anint 3/5=1; dx=3a-5a=-2a Es. dx=2a; anint 2/5=0; dx=2a.")
34
Implementazione in fortran Dove s x,sy,sz =dimensioni della cella di simulazione (solo per celle in cui x,y, e z sono direzioni perpendicolari, se no è più complicato). Notare: può essere cambiato il segno (ed è giusto che sia così, pensare alle forze) di dx,dy,dz. sx=17.64739=sy sz=24.957179
 di dx,dy,dz. sx= =sy sz=")
35
Dimensioni della cella e cutoff rcrc

36
Gli atomi interagiscono con le proprie immagini. Posso usare la minimum image convention per ovviare a questo fatto, ma non è formalmente un gran chè (di fatto introduco un nuovo cutoff). Invece, se L>2r c la minimum image convention viene soddisfatta in modo naturale. Nel caso in figura, la cella di simulazione scelta era troppo piccola. La condizione L>2r c va considerata come condizione minimale per effettuare una simulazione decente.
. Invece, se L>2r c la minimum image convention viene soddisfatta in modo naturale. Nel caso in figura, la cella di simulazione scelta era troppo piccola. La condizione L>2r c va considerata come condizione minimale per effettuare una simulazione decente..")
37
Attenzione! Anche se L>2r c l’utilizzo delle PBC non equivale in tutto e per tutto a simulare un sistema infinito. In un vero sistema infinito ….

40
Nel sistema “replicato” invece …..

43
Tipici problemi connessi ….. Esempio: consideriamo un difetto che provoca un campo di deformazione a lungo raggio. Se la cella con PBC è troppo piccola..

45
E’ chiaro che non posso descrivere una situazione simile con le PBC che forzano uguale distorsione in ogni cella immagine. Con le PBC non sto simulando l’effetto di un difetto isolato ma di un certo coverage di difetti disposti ordinatamente nel sistema

46
Se lo simulo con le PBC e la cella piccola: a)il campo di deformazione sarà diverso da quello vero b)un difetto risentirà del campo di deformazione creato dalla sua immagine (“interazione” laterale spuria). In generale: “impossibilià di simulare un difetto (o un adsorbato) isolato”
 isolato .")
47
Coppia di difetti che si annichilano se si incontrano (interstiziale vacanza)
")
48
Se il difetto si muove e se muovendosi crea un “antidifetto” (potrebbe essere una coppia vacanza/interstiziale)
")
50
la probabilità di annichilazione è molto maggiore di quella reale

51
Notare infine che la densità atomica volumetrica è fissata da N/V della cella principale: non posso simulare fenomeni come l’espansione termica. Se gli atomi al centro della cella aumentano la loro distanza, necessariamente la distanza tra atomi dovrebbe diminuire. Il sistema preferisce allora mantenere distanze uguali

52
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi:

53
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi:

54
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi:

55
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi:

56
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi:

57
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi:

58
Nei codici, ovviamente, non vengono allocate le posizioni di tutte le immagini: la pittura delle celle ripetute non deve ingannare. Se ho processi diffusivi si può verificare la seguente situazione quando visualizzo la posizione degli atomi: Se voglio calcolare il coefficiente di diffusione del’oggetto che si muove (proporzionale allo scarto quadratico medio dalla posizione iniziale), devo considerare la traiettoria vera. Contemporaneamente però, è comodo visualizzare anche quella riferita alla cella principale
, devo considerare la traiettoria vera. Contemporaneamente però, è comodo visualizzare anche quella riferita alla cella principale.")
65
E’ comodo tenere in memoria sia le coordinate reali sia quelle riferite alla sola cella principale di simulazione.

Presentazioni simili





























 ad ogni istante di tempo t sono v.a. Notazione: X : insieme di.>")




