Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
GESTIONE DELLA TECNOLOGIA SANITARIA ANNO 2011-2012
PROF. PAOLO DONATI

2
PROGRAMMA DEL CORSO (I)
INTRODUZIONE: l’oggetto, i soggetti, i ruoli IL SOFTWARE GESTIONALE DEFINIZIONE DI BASE: apparecchiature elettromedicali, dispositivi medici, tecnologie biomediche LA NORMATIVA EUROPEA SUI DISPOSITIVI MEDICI(93/42, 2007/47, i decreti legislativi): finalità e struttura. Definizioni, classificazione dei D.M., procedure di conformità LA CODIFICA NAZIONALE DEI DISPOSITIVI MEDICI (CND)
: finalità e struttura. Definizioni, classificazione dei D.M., procedure di conformità. LA CODIFICA NAZIONALE DEI DISPOSITIVI MEDICI (CND)")
3
PROGRAMMA DEL CORSO (II)
ORGANIZZAZIONE DEL S.S.N.: cenni alla Legislazione Nazionale e della Regione Toscana- Le Aziende Sanitarie e la loro struttura organizzativa IL RISCHIO CLINICO E TECNOLOGICO ACCREDITAMENTO E QUALITÀ LE MODALITÀ DI GESTIONE DELLE TECNOLOGIE IL SERVIZIO DI INGEGNERIA CLINICA: compiti e dimensionamento MODALITÀ E PROCESSI HTA Acquisizione, Accettazione-Collaudo-Installazione e Inventariazione Le Manutenzioni: Correttive, Preventive, Controlli di qualità Il fuori Uso PROCEDURE GESTIONALI E OPERATIVE

4
PROGRAMMA DEL CORSO (III)
MODULO “SICUREZZA ELETTRICA” (Prof. Piccinocchi) APPROFONDIMENTI OSSERVATORIO DELLE TECNOLOGIE SANITARIE: prezzi di acquisto, costi di manutenzione: esempio dell’Osservatorio della Toscana L’INVENTARIO DEI BENI MOBILI IL DIMENSIONAMENTO DI UN SERVIZIO DI INGEGNERIA CLINICA APPLICATO ALLA ASL DI VIBO VALENTIA ALTRO
 APPROFONDIMENTI. OSSERVATORIO DELLE TECNOLOGIE SANITARIE: prezzi di acquisto, costi di manutenzione: esempio dell’Osservatorio della Toscana. L’INVENTARIO DEI BENI MOBILI. IL DIMENSIONAMENTO DI UN SERVIZIO DI INGEGNERIA CLINICA APPLICATO ALLA ASL DI VIBO VALENTIA. ALTRO.")
5
Tutto il materiale è reperibile al sito:
MATERIALE DEL CORSO LUCIDI 2012 (*) DOCUMENTI DI APPROFONDIMENTO DIRETTIVE E LEGISLAZIONE Tutto il materiale è reperibile al sito: (*)I lucidi sono in corso di aggiornamento e verranno caricati sul sito indicato prima delle relative lezioni.
 DOCUMENTI DI APPROFONDIMENTO. DIRETTIVE E LEGISLAZIONE. Tutto il materiale è reperibile al sito: (*)I lucidi sono in corso di aggiornamento e verranno caricati sul sito indicato prima delle relative lezioni.")
6
INTRODUZIONE

7
INTRODUZIONE 1 Ruolo Strategico assunto dalle Tecnologie Sanitarie nella produzione di servizi sanitari siano essi di Prevenzione, Diagnosi, Cura, Riabilitazione. Nell’arco di 20 anni il loro numero è almeno quintuplicato ma soprattutto sono profondamente cambiate le caratteristiche tecnologiche per rispondere a nuove necessità professionali del medico. Si è imposta una nuova figura: l’ingegnere addetto alla gestione delle Tecnologie Sanitarie in ogni fase del processo: l’ingegnere clinico.

8
L’ INGEGNERE CLINICO USO SICURO: USO APPROPIATO ECONOMICO:
Partecipa in modo diretto al processo di GESTIONE DEL RISCHIO in merito alle apparecchiature e deve garantire un uso SICURO, APPROPIATO ed ECONOMICO delle tecnologie sanitarie. USO SICURO: Attività Programmate (VS-CQ-MP) Gestione del rischio Pianificazione sostituzioni Formazione USO APPROPIATO ECONOMICO: Ottimizzazione Acquisti Mantenimento standard di qualità e sicurezza
 Gestione del rischio. Pianificazione sostituzioni. Formazione. USO APPROPIATO ECONOMICO: Ottimizzazione Acquisti. Mantenimento standard di qualità e sicurezza.")
9
Ma oggi è sufficiente tutto ciò o siamo oltre?
INTRODUZIONE 1.1 Ma oggi è sufficiente tutto ciò o siamo oltre? Non è più possibile parlare di tecnologia se non congiuntamente all’ICT. Quindi: Ingegnere clinico più informatico, più ing. esperto di reti e telecomunicazioni. Ma ancora: L’ospedale è una struttura complessa e quindi: la progettazione e gli impianti. Quindi una visione interdisciplinare

10
INTRODUZIONE 2 Non stiamo dimenticando una figura? ……
Il ruolo strategico della Tecnologia in senso lato ha imposto e richiede oggi a chi ne ha la responsabilità (Chi? lo vedremo) di compiere scelte strategiche, organizzative e gestionali (strutture interne o affidate all’esterno), di dotarsi di procedure (accreditamento e qualità), di strumenti di controllo e monitoraggio (software), di strumentazione se si è scelto di svolgere in proprio una parte della attività, ecc. Iniziamo il corso con uno strumento concreto di governo e gestione delle Apparecchiature elettromedicali: il Software Metis. E’ come un indice.
 di compiere scelte strategiche, organizzative e gestionali (strutture interne o affidate all’esterno), di dotarsi di procedure (accreditamento e qualità), di strumenti di controllo e monitoraggio (software), di strumentazione se si è scelto di svolgere in proprio una parte della attività, ecc. Iniziamo il corso con uno strumento concreto di governo e gestione delle Apparecchiature elettromedicali: il Software Metis. E’ come un indice.")
11
SOFTWARE GESTIONALE

13
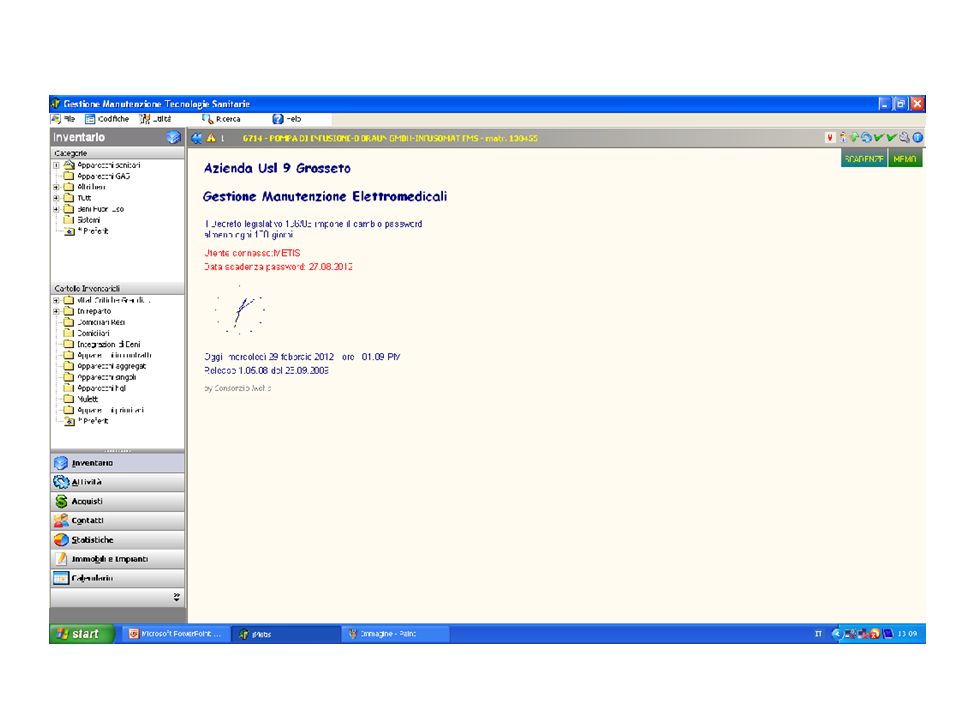
IL software gestionale 1
Consente il “governo” delle Tecnologie Sanitarie nelle differenti fasi: programmazione, procedure di acquisizione, ingresso in Azienda, manutenzione-sicurezza-qualità, dismissione. . Consente di monitorare la vita dell’Apparecchiatura: la sua storia manutentiva Consente di compiere valutazioni tecnico economiche ed assicurare un uso appropriato e sicuro Diventeranno familiari termini quali: Collaudo Inventario Manutenzione preventiva o a programma Manutenzione correttiva Verifiche di Sicurezza Libro Macchina Fuori Uso Azienda USL e Ospedaliera, Unità Operativa, Dipartimento, zona, Centro di responsabilità, centro di costo, ecc.

15
IL SOFTWARE GESTIONALE 2
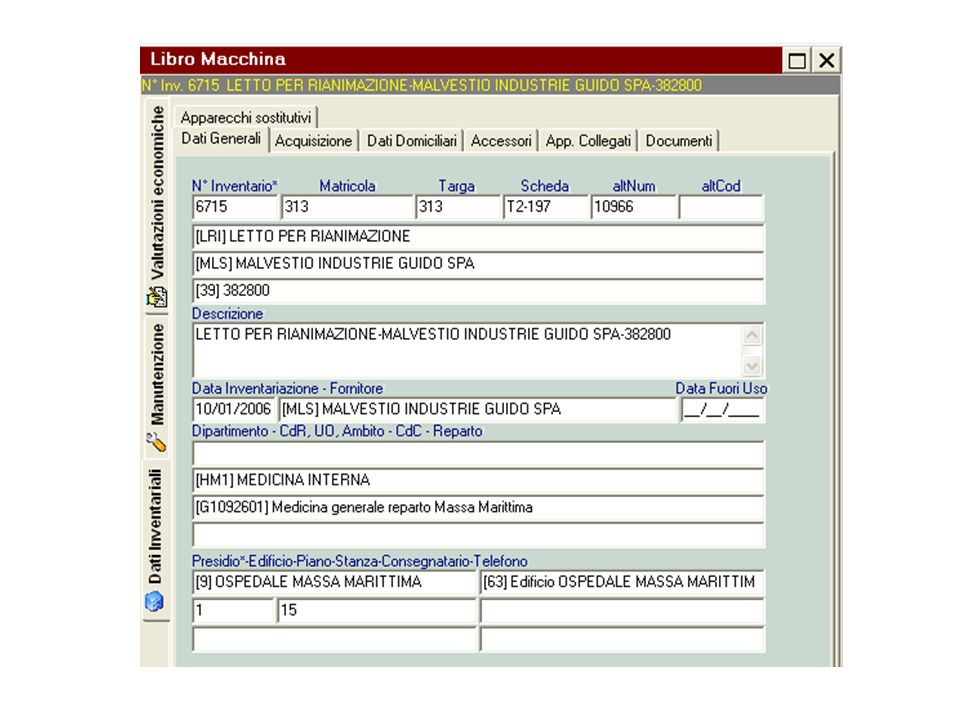
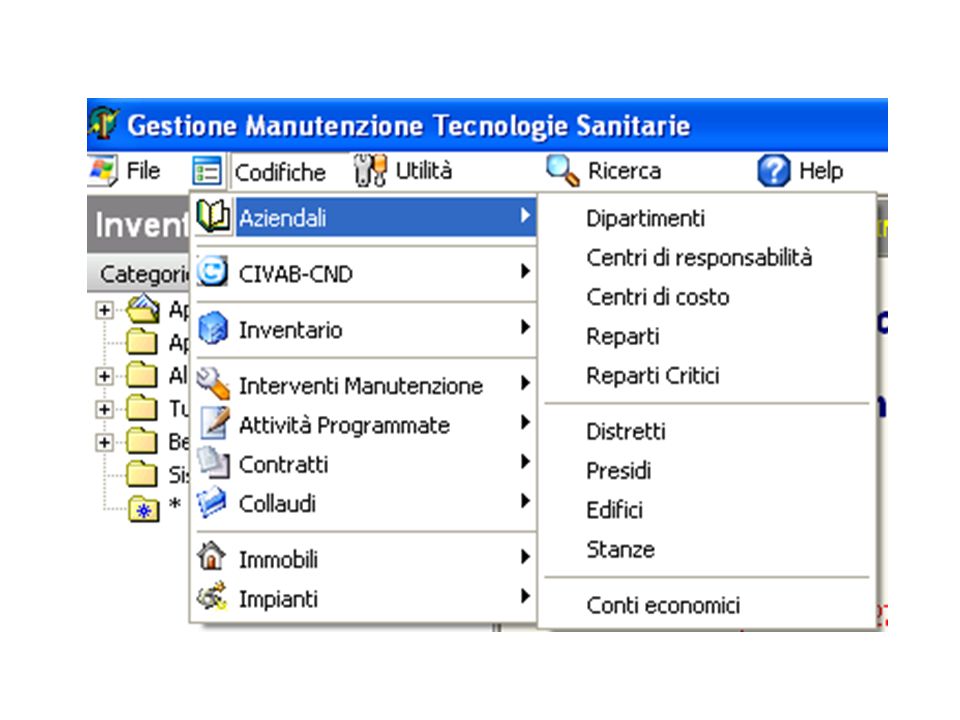
Alla base della progettazione del Software vi è una “analisi dei requisiti” che individua gli “oggetti” ed i “processi”, i “soggetti” coinvolti a livello di persone e di strutture organizzative. Punto centrale è il Bene (apparecchiatura elettromedicale) con le sue caratteristiche tecniche; entra a far parte del Patrimonio Aziendale (non sempre), ha una collocazione fisica; chi ne è responsabile ingegnere o medico? (Quale risposta date? La domanda è posta correttamente?); chi sono gli utilizzatori, quali i processi? Il Software ha una barra principale con le seguenti voci: Codifica Manutenzione Utilità Perché una codifica e di che cosa?
 con le sue caratteristiche tecniche; entra a far parte del Patrimonio Aziendale (non sempre), ha una collocazione fisica; chi ne è responsabile ingegnere o medico (Quale risposta date La domanda è posta correttamente ); chi sono gli utilizzatori, quali i processi Il Software ha una barra principale con le seguenti voci: Codifica. Manutenzione. Utilità. Perché una codifica e di che cosa")
17
Il software gestionale 3
CODIFICA : consente di identificare in modo univoco gli oggetti Per le Apparecchiature: AcMaGest (CIVAB) Codifica nazionale, la sua struttura è caratterizzata da 3 caratteri alfabetici per la classe, 3 caratteri alfabetici per la ditta, 2 caratteri alfanumerici per il modello. NUOVA CODOFICA MINISTERO SALUTE (CND). Altre GMDN, ECRI. (vedere lucidi di approfondimento) I dettagli delle altre codifiche le commentiamo nell’analisi del software: le voci principali: Contratti ; Componenti funzionali : Interventi ; Collegate al bene, Norme CEI 62-5 ;Magazzino ANAGRAFICA: significa individuare in modo univoco le specificazioni dei luoghi, dell’organizzazione, dei soggetti coinvolti. Vediamo e commentiamo dal software Bene: fondamentale l’anagrafica inventariale; Ditte ; Strutture Aziendali ; Varie
 Codifica nazionale, la sua struttura è caratterizzata da 3 caratteri alfabetici per la classe, 3 caratteri alfabetici per la ditta, 2 caratteri alfanumerici per il modello. NUOVA CODOFICA MINISTERO SALUTE (CND). Altre GMDN, ECRI. (vedere lucidi di approfondimento) I dettagli delle altre codifiche le commentiamo nell’analisi del software: le voci principali: Contratti ; Componenti funzionali : Interventi ; Collegate al bene, Norme CEI 62-5 ;Magazzino. ANAGRAFICA: significa individuare in modo univoco le specificazioni dei luoghi, dell’organizzazione, dei soggetti coinvolti. Vediamo e commentiamo dal software. Bene: fondamentale l’anagrafica inventariale; Ditte ; Strutture Aziendali ; Varie.")
18
LA CODIFICA CIVAB XXX JJJ 3Y ECT TOS E4
La codifica CIVAB rappresenta un sistema univoco di riconoscimento di una parte consistente delle tecnologie biomediche presenti sul mercato nazionale. Il codice è costituito da una stringa di 8 caratteri alfanumerici attraverso la quale si individuano: La Classe di tecnologia (primi tre caratteri Alfabetici) (es: ECT = ECOTOMOGRAFO) La Ditta Produttrice (seconda terna di caratteri Alfabetici) (es: SIE = Siemens ) Lo specifico Modello di tecnologia di quella classe e di quel produttore (ultimi due caratteri Alfnumerici) 877 Classi 3417 Ditte 53266 Modelli XXX JJJ Y ECT TOS E4 Es. Classe Modello ECOTOMOGRAFO SSA 550 A E4 NEMIO PREMIUM Produttore TOSHIBA CORP MEDICAL SYSTEMS
 (es: ECT = ECOTOMOGRAFO) La Ditta Produttrice (seconda terna di caratteri Alfabetici) (es: SIE = Siemens ) Lo specifico Modello di tecnologia di quella classe e di quel produttore (ultimi due caratteri Alfnumerici) 877 Classi Ditte Modelli. XXX JJJ 3Y. ECT TOS E4. Es. Classe. Modello. ECOTOMOGRAFO. SSA 550 A E4 NEMIO PREMIUM. Produttore. TOSHIBA CORP MEDICAL SYSTEMS.")
19
Il software gestionale 4
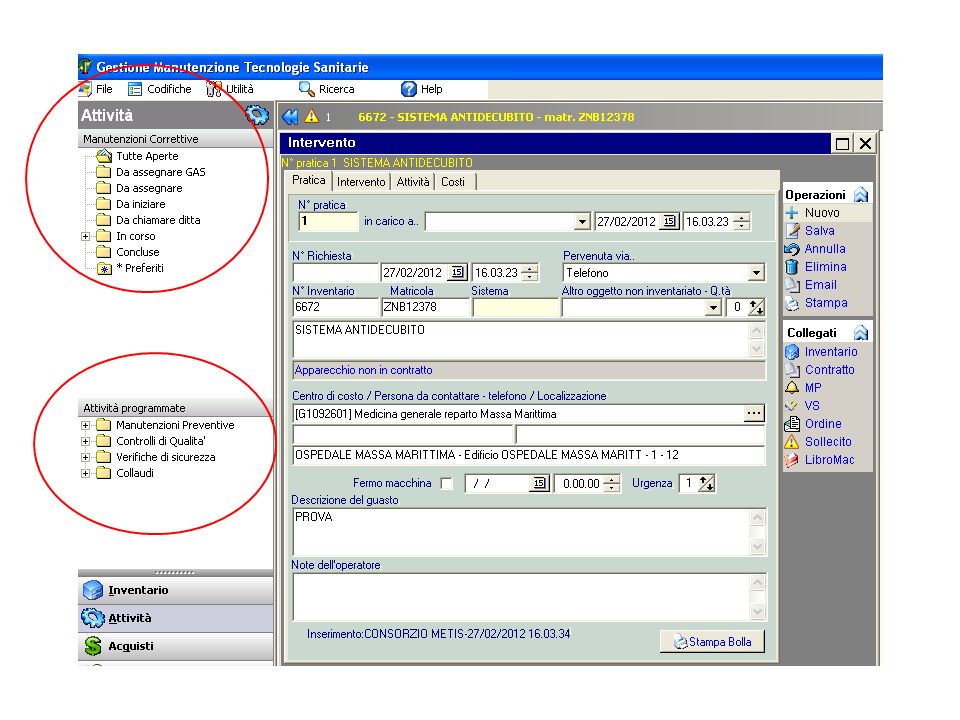
MANUTENZIONE: una delle principali funzioni tra le attività delle U.O.T.S e di qualunque altro soggetto incaricato, anche esterno. Intervento ; Schede Tecniche Contratti Listini ; Magazzino Verifiche di Sicurezza Manutenzione Preventiva Fermo Macchina Collaudi UTILITA’ Gestioni Acquisti Report Budget

21
DEFINIZIONI DI BASE

22
TECNOLOGIE BIOMEDICHE APPARECCHIATURE ELETTROMEDICALI
CERCHIAMO DI CAPIRE LE DIFFERENZE… TECNOLOGIE BIOMEDICHE DISPOSITI MEDICI APPARECCHIATURE ELETTROMEDICALI

23
TECNOLOGIE BIOMEDICHE APPARECCHIATURE ELETTROMEDICALI
OMS: Tutti gli strumenti, apparecchiature, farmaci e procedure impiegati nella erogazione dei servizi sanitari, nonché i sistemi organizzativi e di supporto attraverso i quali l’assistenza sanitaria viene fornita. MdS: L’insieme dei prodotti e dei dispositivi medici che afferiscono al settore della sanità ad eccezione dei farmaci; le apparecchiature biomediche costituiscono un sotto insieme di tale comparto, con riferimento alla sola strumentazione. DISPOSITIVO MEDICO CEE 93/42: “ uno strumento,un apparecchio, un impianto, una sostanza, o altro prodotto usato da solo o in combinazione, compreso il software informatico impiegato per il corretto funzionamento, e destinato dal fabbricante ad essere impiegato nell’uomo a scopo di: diagnosi, prevenzione, controllo, terapia, o attenuazione di una malattia diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un handicap, studio, sostituzione o modifica dell’anatomia o di un processo fisiologico, intervento sul concepimento purché non eserciti l’azione principale nel o sul corpo umano, cui è destinato, con mezzi farmacologici o immunologici, né mediante processo metabolico, ma la cui funzione possa essere coadiuvata da tali mezzi.” APPARECCHIATURE ELETTROMEDICALI CEI 62.5: Apparecchio elettrico, munito di non più di una connessione a una particolare rete di alimentazione, destinato alla diagnosi, al trattamento o alla sorveglianza del paziente sotto la super visione del medico, e che entra in contatto fisico o elettrico col paziente e/o trasferisce energia verso o dal paziente e/o rivela un determinato trasferimento di energia verso o dal paziente.

24
Tecnologie Biomediche
Classificazione delle Tecnologie Biomediche in base alla loro funzione Tecnologie Biomediche Area Diagnostica Area Terapeutica-Riabilitativa Valutazione Funzionale Bioimmagini Diagnostica clinica Terapia Chirurgica Interventistica a bassa invasività Terapia non invasiva Organi artificiali, protesi Riabilitazione Supporto

25
Valutazione funzionale

26
Bioimmagini

27
Terapia Chirurgica

28
Interventistica a bassa invasività

29
Terapia non invasiva

30
Organi artificiali e protesi

31
Riabilitazione e Supporto

32
DIRETTIVA 93/42 2007/47 I DISPOSITIVI MEDICI

33
DIRETTIVA EUROPEA 93/42 (RECEPITA IN ITALIA CON DLGS 46/97)
Regolamenta su base comunitaria l’immissione in commercio dei Dispositivi Medici. La conformità dei prodotti viene dimostrata dalla presenza del marchio CE Il marchio CE deve essere affisso in maniera indelebile e leggibile sui dispositivi, sulle istruzioni d’uso e sulle confezioni di vendita.

34
IL NUOVO APPROCCIO Il ‘Nuovo Approccio’ si differenzia dallo schema tradizionale perché garantisce che i prodotti di tutti i Paesi dell’Unione Europea rispondano agli stessi requisiti essenziali e che siano individuate delle ‘Autorità Competenti’ che effettuino azioni di Sorveglianza.

35
DALLA 93/42 ALLA 2007/47 La nuova direttiva sui Dispositivi Medici, emanata il 5 settembre del 2007 e recepita il Italia a marzo 2010, con il Decreto Legislativo 37/2010, rappresenta il risultato di un importante lavoro di mediazione fra gli Stati membri dell’Unione Europea e introduce delle sostanziali modifiche alle precedenti direttive. La nuova direttiva realizza la convergenza con la direttiva comunitaria relativa ai dispositivi impiantabili attivi (90/385) e l’armonizzazione con la direttiva per il trattamento del sangue e suoi derivati (2002/98) ed entra nel merito di alcune problematiche che la direttiva precedente (93/42) non aveva approfondito.
 e l’armonizzazione con la direttiva per il trattamento del sangue e suoi derivati (2002/98) ed entra nel merito di alcune problematiche che la direttiva precedente (93/42) non aveva approfondito.")
36
UN IMPORTANTE MODIFICA
Nuova Definizione di Dispositivo Medico 2007/47 (DLGS 37/2010) “ uno strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compresi gli accessori tra cui il software, destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo stesso, destinato dal fabbricante ad essere impiegato sull’uomo a fini di (…).” 93/47 (DLGS 46/97) “uno strumento,un apparecchio, un impianto, una sostanza, o altro prodotto usato da solo o in combinazione, compreso il software informatico impiegato per il corretto funzionamento, e destinato dal fabbricante ad essere impiegato nell’uomo a scopo di (…)
 uno strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compresi gli accessori tra cui il software, destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo stesso, destinato dal fabbricante ad essere impiegato sull’uomo a fini di (…). 93/47 (DLGS 46/97) uno strumento,un apparecchio, un impianto, una sostanza, o altro prodotto usato da solo o in combinazione, compreso il software informatico impiegato per il corretto funzionamento, e destinato dal fabbricante ad essere impiegato nell’uomo a scopo di (…)")
37
ALCUNE DEFINIZIONI(1) DISPOSITIVO MEDICO
qualunque strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compreso il software destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche o terapeutiche e necessario al corretto funzionamento del dispositivo, destinato dal fabbricante ad essere impiegato sull'uomo a fini di diagnosi, prevenzione, controllo, terapia o attenuazione di una malattia; di diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un handicap; di studio, sostituzione o modifica dell'anatomia o di un processo fisiologico; di intervento sul concepimento, il quale prodotto non eserciti l'azione principale, nel o sul corpo umano, cui è destinato, con mezzi farmacologici o immunologici né mediante processo metabolico ma la cui funzione possa essere coadiuvata da tali mezzi. ACCESSORIO prodotto che, pur non essendo un dispositivo, sia destinato in modo specifico dal fabbricante ad essere utilizzato con un dispositivo per consentirne l'utilizzazione prevista dal fabbricante stesso.

38
DISPOSITIVO DI DIAGNOSI IN VITRO DISPOSITIVI PER INDAGINI CLINICHE
ALCUNE DEFINIZIONI(2) DISPOSITIVO DI DIAGNOSI IN VITRO qualsiasi dispositivo composto da un reagente, da un prodotto reattivo, da un insieme, da uno strumento, da un apparecchio o da un sistema utilizzato da solo o in combinazione, destinato dal fabbricante ad essere impiegato in vitro per l'esame di campioni provenienti dal corpo umano al fine di fornire informazioni sugli stati fisiologici o sugli stati sanitari o di malattia o anomalia congenita. DISPOSITIVO SU MISURA qualsiasi dispositivo fabbricato appositamente, sulla base della prescrizione scritta di un medico debitamente qualificato e indicante, sotto la responsabilità del medesimo, le caratteristiche specifiche di progettazione del dispositivo e destinato ad essere utilizzato solo per un determinato paziente. La prescrizione può essere redatta anche da altra persona la quale vi sia autorizzata in virtù della propria qualificazione professionale. I dispositivi fabbricati con metodi di fabbricazione continua od in serie, che devono essere successivamente adattati, per soddisfare un'esigenza specifica del medico o di un altro utilizzatore professionale, non sono considerati dispositivi su misura. DISPOSITIVI PER INDAGINI CLINICHE un dispositivo destinato ad essere messo a disposizione di un medico debitamente qualificato per lo svolgimento di indagini di cui all'allegato X, punto 2.1, in un ambiente clinico umano adeguato. Per l'esecuzione delle indagini cliniche, al medico debitamente qualificato è assimilata ogni altra persona, la quale, in base alla propria qualificazione professionale, sia autorizzata a svolgere tali indagini.
 DISPOSITIVO DI DIAGNOSI IN VITRO. qualsiasi dispositivo composto da un reagente, da un prodotto reattivo, da un insieme, da uno strumento, da un apparecchio o da un sistema utilizzato da solo o in combinazione, destinato dal fabbricante ad essere impiegato in vitro per l esame di campioni provenienti dal corpo umano al fine di fornire informazioni sugli stati fisiologici o sugli stati sanitari o di malattia o anomalia congenita. DISPOSITIVO SU MISURA. qualsiasi dispositivo fabbricato appositamente, sulla base della prescrizione scritta di un medico debitamente qualificato e indicante, sotto la responsabilità del medesimo, le caratteristiche specifiche di progettazione del dispositivo e destinato ad essere utilizzato solo per un determinato paziente. La. prescrizione può essere redatta anche da altra persona la quale vi sia autorizzata in virtù della propria qualificazione professionale. I dispositivi fabbricati con metodi di fabbricazione continua od in serie, che devono essere successivamente adattati, per soddisfare un esigenza specifica del medico o di un altro utilizzatore professionale, non sono considerati dispositivi su misura. DISPOSITIVI PER INDAGINI CLINICHE. un dispositivo destinato ad essere messo a disposizione di un medico debitamente qualificato per lo svolgimento di indagini di cui all allegato X, punto 2.1, in un ambiente clinico umano adeguato. Per l esecuzione delle indagini cliniche, al medico debitamente qualificato è assimilata ogni altra persona, la quale, in base alla propria qualificazione professionale, sia autorizzata a svolgere tali indagini.")
39
IMMISSIONE IN COMMERCIO
ALCUNE DEFINIZIONI(3) DESTINAZIONE L'utilizzazione alla quale è destinato il dispositivo secondo le indicazioni fornite dal fabbricante nell'etichetta, nel foglio illustrativo o nel materiale Pubblicitario. IMMISSIONE IN COMMERCIO La prima messa a disposizione a titolo oneroso o gratuito di dispositivi, esclusi quelli destinati alle indagini cliniche, in vista della distribuzione o utilizzazione sul mercato comunitario, indipendentemente dal fatto che si tratti di dispositivi nuovi o rimessi a nuovo. (ART. 3 :I dispositivi possono essere immessi in commercio o messi in servizio unicamente se rispondono ai requisiti prescritti dal presente decreto, sono correttamente forniti e installati, sono oggetto di un’adeguata manutenzione e sono utilizzati in conformità della loro destinazione). MESSA IN SERVIZIO Fase in cui il dispositivo è stato reso disponibile all’utilizzatore finale in quanto pronto per la prima utilizzazione sul mercato comunitario secondo la sua destinazione d’uso.
 DESTINAZIONE. L utilizzazione alla quale è destinato il dispositivo secondo le indicazioni fornite dal fabbricante nell etichetta, nel foglio illustrativo o nel materiale Pubblicitario. IMMISSIONE IN COMMERCIO. La prima messa a disposizione a titolo oneroso o gratuito di dispositivi, esclusi quelli destinati alle indagini cliniche, in vista della distribuzione o utilizzazione sul mercato comunitario, indipendentemente dal fatto che si tratti di dispositivi nuovi o rimessi a nuovo. (ART. 3 :I dispositivi possono essere immessi in commercio o messi in servizio unicamente se rispondono ai requisiti prescritti dal presente decreto, sono correttamente forniti e installati, sono oggetto di un’adeguata manutenzione e sono utilizzati in conformità della loro destinazione). MESSA IN SERVIZIO. Fase in cui il dispositivo è stato reso disponibile all’utilizzatore finale in quanto pronto per la prima utilizzazione sul mercato comunitario secondo la sua destinazione d’uso.")
40
I RESPONSABILI DELL’IMMISSIONE IN COMMERCIO
il Fabbricante: soggetto che si assume la responsabilità della conformità del prodotto, ma che può non esserne il produttore materiale, potendo affidare a terzi la realizzazione dello stesso o anche solo una parte del processo produttivo. Il fabbricante è la figura che deve accertare che il suo prodotto soddisfi i requisiti essenziali di sicurezza ed efficacia predisponendo un dossier tecnico che includa tutta la documentazione relativa alla progettazione, gestione dei rischi, dati clinici che ne confermino l’efficacia, e indicazioni sulle procedure di sorveglianza nella fase successiva all’immissione in commercio (post-marketing), comprendenti la rintracciabilità, le segnalazione di incidenti e il ritiro dal commercio (allegato VII D.Lgs. 46/97). Il Mandatario del Fabbricante: figura designata dal Fabbricante non comunitario per rappresentarlo o agire in sua vece. Deve aver sede in uno dei Paesi dell’Unione Europea ed è il riferimento per le Autorità competenti.
, comprendenti la rintracciabilità, le segnalazione di incidenti e il ritiro dal commercio (allegato VII D.Lgs. 46/97). Il Mandatario del Fabbricante: figura designata dal Fabbricante non comunitario per rappresentarlo o agire in sua vece. Deve aver sede in uno dei Paesi dell’Unione Europea ed è il riferimento per le Autorità competenti.")
41
I REQUISITI ESSENZIALI
I requisiti essenziali (Allegato I) includono tutte le caratteristiche dei dispositivi medici di qualsiasi tipologia che possono incidere sulla sicurezza ed efficacia in relazione all’uso finale stabilito dal fabbricante e che sono indispensabili per ottenere il livello di protezione atteso. REQUSITI ESSENZIALI GENERALI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (rivolti agli aspetti tecnologici)
 includono tutte le caratteristiche dei dispositivi medici di qualsiasi tipologia che possono incidere sulla sicurezza ed efficacia in relazione all’uso finale stabilito dal fabbricante e che sono indispensabili per ottenere il livello di protezione atteso. REQUSITI ESSENZIALI. GENERALI. RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (rivolti agli aspetti tecnologici)")
42
I REQUISITI GENERALI (1)
")
43
I REQUISITI GENERALI (2)
")
44
I REQUISITI GENERALI (3-6)
")
45
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (7)
")
46
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (8)
")
47
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (9)
")
48
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (11)
")
49
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (12)
")
50
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (13)
")
51
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (13)
")
52
I REQUISITI RELATIVI ALLA PROGETTAZIONE E COSTRUZIONE (13)
52

53
IL FASCICOLO TECNICO Il Fascicolo Tecnico deve avere la seguente struttura: Descrizione del Prodotto Schemi di progettazione e metodi di fabbricazione Risultati dell’analisi del rischio Norme tecniche applicate Tabella di rispondenza ai requisiti essenziali Relazioni di prova e ove necessario i dati clinici di cui all’allegato X Progetto di etichettatura e istruzioni d’uso

54
IL MANUALE D’USO Il manuale d’uso o foglietto illustrativo, oltre alle indicazioni previste per l’etichettatura, deve anche riportare: Prestazioni ed effetti collaterali Informazioni sulla connessione con altri dispositivi Installazione e manutenzione Rischi connessi con l’impianto di un dispositivo Rischi di interferenze reciproche, durante indagini o trattamenti specifici Risterilizzazione in caso di danneggiamento dell’involucro Trattamenti appropriati per i dispositivi per i quali è prevista la riutilizzazione Informazioni su trattamenti o manipolazioni specifiche Informazioni per dispositivi che emettono radiazioni Altre indicazioni per operatori sanitari ai fini di adottare le corrette precauzione nell’uso del dispositivo.

55
ALTRE IMPORTANTI MODIFICHE DELLA 2007/47
Nuovi requisiti per la Valutazione Clinica e Riduzione dei RISCHI Si è posta in risalto la necessità della riduzione dei rischi di errore nell’uso del dispositivo, ponendo l’accento sul livello di formazione e delle conoscenze degli utilizzatori e la previsione di una valutazione clinica di tutti i dispositivi medici per la dimostrazione dei requisiti essenziali ed in particolare dell’efficacia clinica. Una mancata efficacia di un dispositivo medico può tradursi in un aumento del rischio della sicurezza del paziente. Il fabbricante ha l’obbligo di dimostrare che i propri prodotti forniscono le prestazioni ad essi attribuite, in modo da poter espletare una o più funzioni che caratterizzano i dispositivi medici (attività terapeutica, preventiva, diagnostica…). Viene richiesto che siano presenti dati clinici sia di tipo bibliografico che derivanti da sperimentazioni ad hoc che il fabbricante è tenuto ad effettuare per una migliore valutazione della sicurezza e dell’efficacia.
. Viene richiesto che siano presenti dati clinici sia di tipo bibliografico che derivanti da sperimentazioni ad hoc che il fabbricante è tenuto ad effettuare per una migliore valutazione della sicurezza e dell’efficacia.")
56
Interrelazioni con altri ambiti normativi (Direttiva Macchine e DPI)
I dispositivi che sono anche macchine, dovranno rispettare i requisiti essenziali stabiliti dalla ‘Direttiva Macchine’, qualora questi siano più specifici di quelli stabiliti dalla direttiva sui Dispositivi Medici, così come i dispositivi ai quali il fabbricante attribuisce anche un’azione di protezione dell’operatore dovranno rispondere ai requisiti essenziali della direttiva sui Dispositivi di Protezione Individuale. Regole di Classificazione La nuova Direttiva modifica alcune classi dei dispositivi, come per gli invasivi chirurgici ad uso temporaneo che vengono in contatto con il sistema nervoso centrale, ora in classe III; dispositivi destinati a disinfettare i dispositivi medici invasivi, ora in classe IIb; i dispositivi attivi destinati a registrare immagine ottenute con i raggi X, ora in classe IIa; i dispositivi che entrano in contatto con l’arco della aorta e con l’aorta discendente fino alla biforcazione, ora in classe III.

57
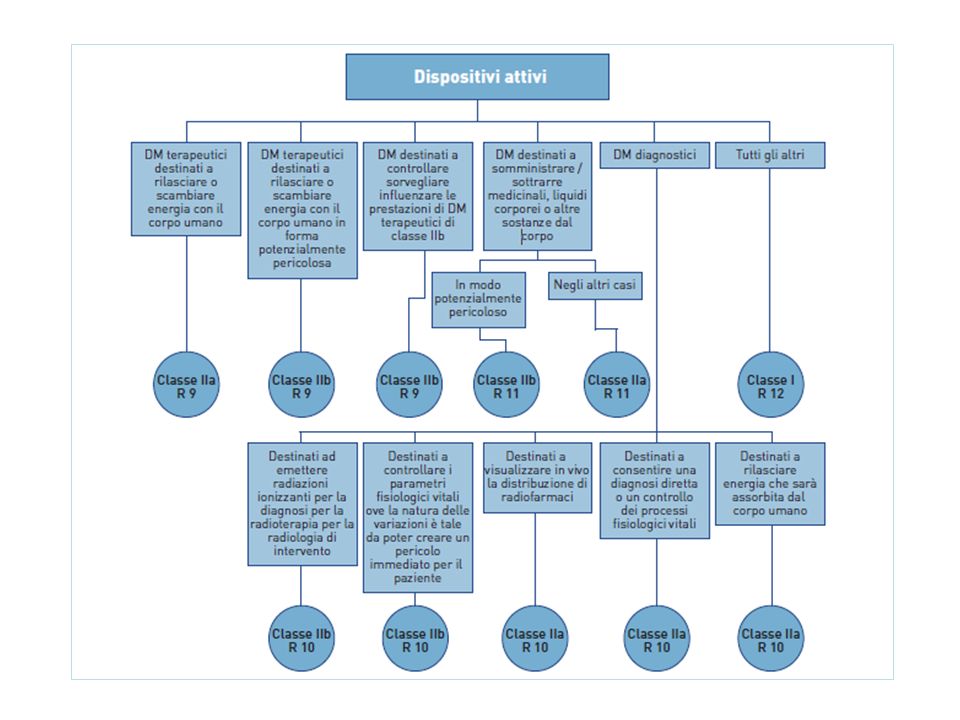
CLASSIFICAZIONE DM Classe I: dispositivi meno critici, quali la gran parte di quelli non attivi e non invasivi, ulteriormente suddivisi in Classe Is : dispositivi forniti allo stato sterile Classe Im: dispositivi che svolgono una funzione di misura; Classe IIa: dispositivi a rischio medio, quali alcuni dispositivi non attivi (invasivi e non) e dispositivi attivi che interagiscono con il corpo in maniera non pericolosa; Classe IIb: dispositivi a rischio medio/alto, quali alcuni dispositivi non attivi (specie invasivi) e i dispositivi attivi che interagiscono con il corpo in maniera pericolosa; Classe III: dispositivi ad alto rischio, quali gran parte dei dispositivi impiantabili, quelli contenenti farmaci o derivanti animali ed alcuni dispositivi che interagiscono sulle funzioni di organi vitali.
 e dispositivi attivi che interagiscono con il corpo in maniera non pericolosa; Classe IIb: dispositivi a rischio medio/alto, quali alcuni dispositivi non attivi (specie invasivi) e i dispositivi attivi che interagiscono con il corpo in maniera pericolosa; Classe III: dispositivi ad alto rischio, quali gran parte dei dispositivi impiantabili, quelli contenenti farmaci o derivanti animali ed alcuni dispositivi che interagiscono sulle funzioni di organi vitali.")
58
durata del contatto con il paziente
REGOLE DI CLASSIFICAZIONE (Allegato IX- sezione I) durata del contatto con il paziente temporanea: < 60 minuti a breve termine: < 30 giorni a lungo termine: > 30giorni sede anatomica sistema circolatorio centrale sistema nervoso invasività non invasivo: non penetra nel corpo, né attraverso un orefizio né attraverso la cute; invasivo tramite orefizi naturali: penetra solo attraverso gli orefizi naturali del corpo (aperture naturali e/o artificiali permanenti); invasivo chirurgico: penetrano nel corpo sia in contesto di intervento che non; impiantabile destinati ad essere impiantati nel corpo. funzionamento non attivo: non ricorrono a fonte di energia; attivo: che ricorrono a qualche forma di energia elettrica o di altro tipo per funzionare (il software stand-alone è considerato un dispositivo medico attivo); attivo terapeutico: destinati a sostenere, modificare, sostituire o ripristinare le funzioni o le strutture biologiche nel contesto di un trattameto o per alleviare una ferita o un handicap; attivo diagnostico: destinato a fornire informazioni riguardanti la diagnosi, il controllo o il trattamento di stati fisiologici di salute, di malattia e di malformazioni.
 durata del contatto con il paziente. temporanea: < 60 minuti. a breve termine: < 30 giorni. a lungo termine: > 30giorni. sede anatomica. sistema circolatorio centrale. sistema nervoso. invasività. non invasivo: non penetra nel corpo, né attraverso un orefizio né attraverso la cute; invasivo tramite orefizi naturali: penetra solo attraverso gli orefizi naturali del corpo (aperture naturali e/o artificiali permanenti); invasivo chirurgico: penetrano nel corpo sia in contesto di intervento che non; impiantabile destinati ad essere impiantati nel corpo. funzionamento. non attivo: non ricorrono a fonte di energia; attivo: che ricorrono a qualche forma di energia elettrica o di altro tipo per funzionare (il software stand-alone è considerato un dispositivo medico attivo); attivo terapeutico: destinati a sostenere, modificare, sostituire o ripristinare le funzioni o le strutture biologiche nel contesto di un trattameto o per alleviare una ferita o un handicap; attivo diagnostico: destinato a fornire informazioni riguardanti la diagnosi, il controllo o il trattamento di stati fisiologici di salute, di malattia e di malformazioni.")
59
REGOLE DI CLASSIFICAZIONE (Allegato IX- sezione II)
Se un dispositivo medico ha più possibilità d’impiego, sarà quello più critico che determinerà la classe. Ugualmente se ad un dispositivo vengono applicate più regole per la classificazione, si utilizzeranno le regole che portano alla classificazione più elevata. Gli accessori sono classificati separatamente dal dispositivo con cui sono impiegati; comunque il software che serve a far funzionare un dispositivo o ad influenzarne l’uso rientra automaticamente nella stessa classe del dispositivo.

60
REGOLE DI CLASSIFICAZIONE (Allegato IX- sezione III)
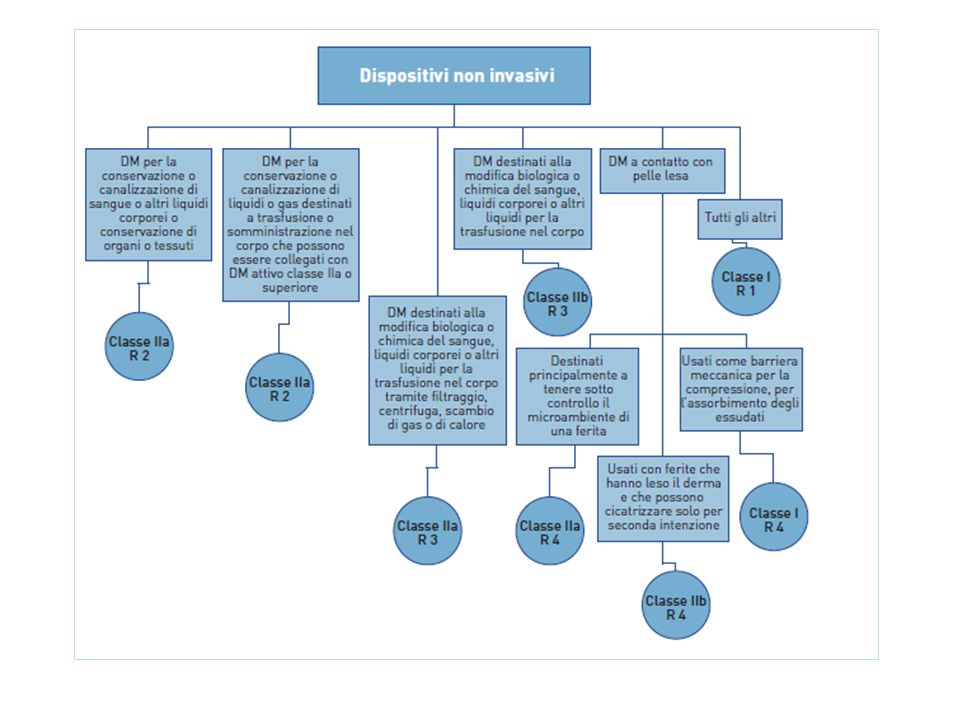
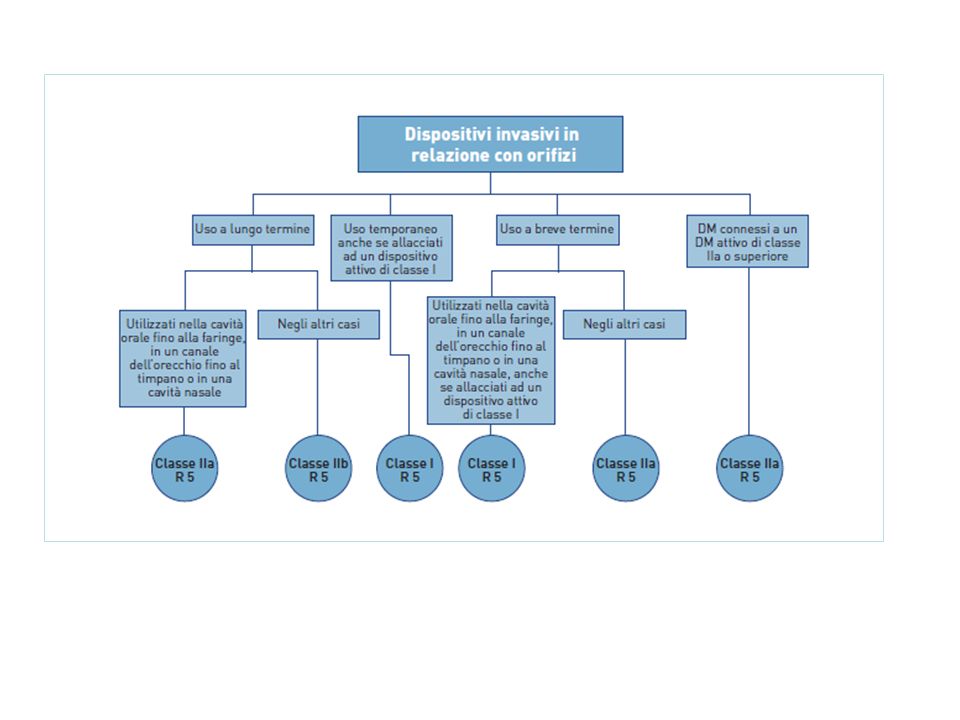
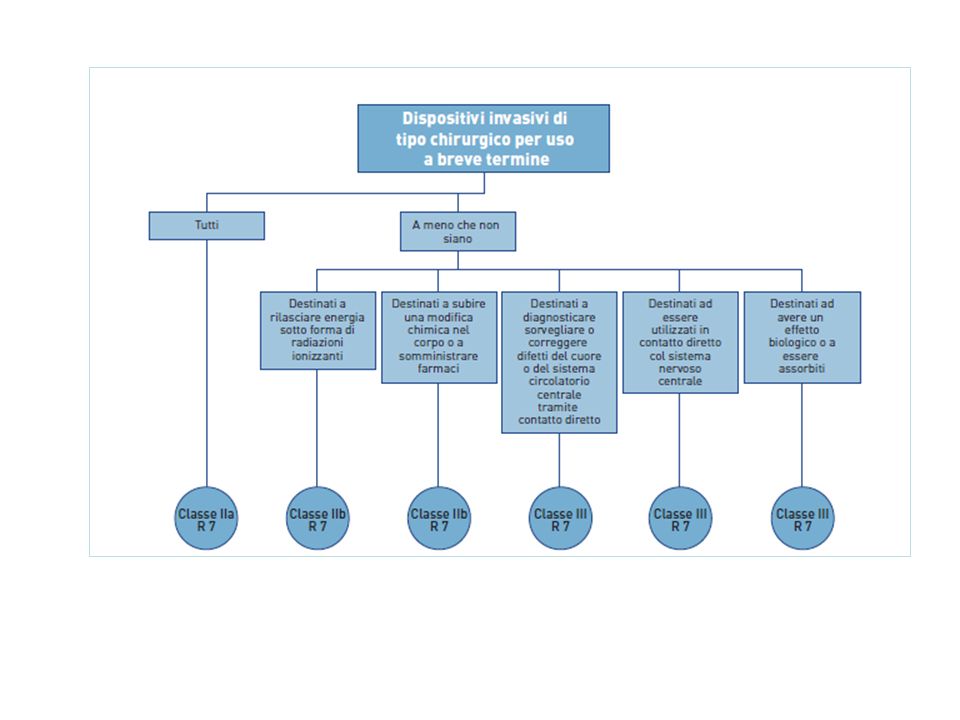
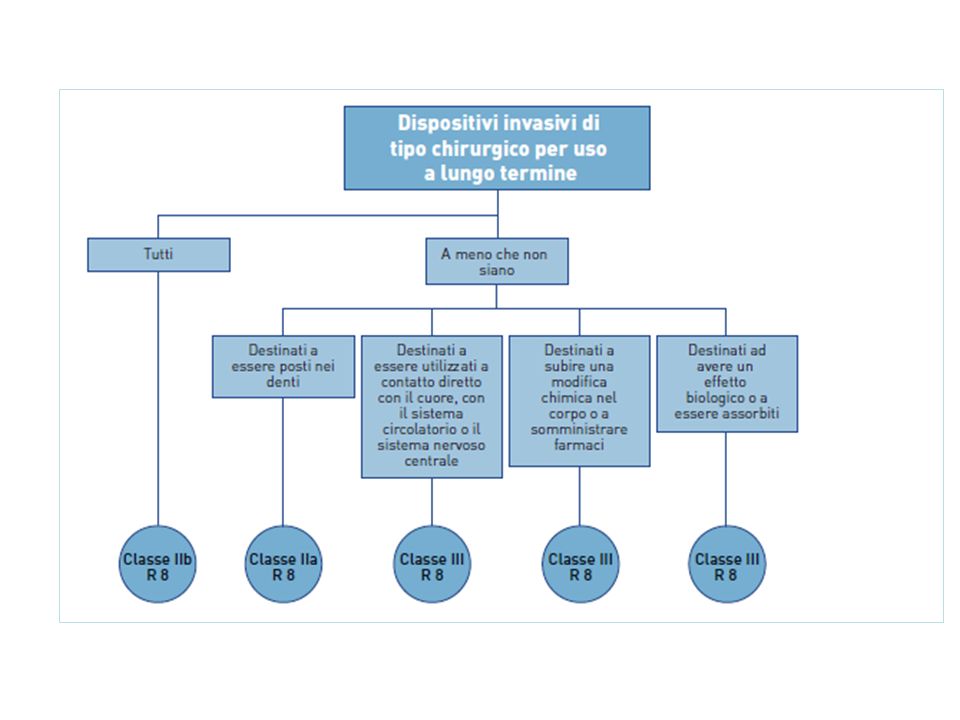
Riporta 19 regole specifiche di classificazione : • Dispositivi non invasivi, 4 regole nn.1, 2, 3, 4 • Dispositivi invasivi, 4 regole nn. 5, 6, 7, 8 • Dispositivi attivi, 4 regole nn. 9, 10, 11,12 • Regole speciali, 7 regole nn. 13, 14, 15, 16, 17, 18, 19

68
PROCEDURE DI CONFORMITÀ
PER TUTTI I DISPOSITIVI La conformità viene valutata, per tutti i dispositivi di qualsiasi classe, dal fabbricante, che la attesta con una dichiarazione detta DICHIARAZIONE DI CONFORMITÀ PER CLASSI > I Per i dispositivi di classe superiore alla I e per alcuni aspetti relativi ai dispositivi di classe Is e Im la conformità viene valutata, con diverse modalità, anche da un soggetto di terza parte, denominato Organismo Notificato, che la attesta mediante una certificazione rilasciata al fabbricante detta CERTIFICATO CE

69
LA DICHIARAZIONE DI CONFORMITÀ
nome e indirizzo del fabbricante o del rappresentante autorizzato che rilascia la dichiarazione; identificazione del prodotto (nome, tipo o numero del modello ed eventuali informazioni supplementari quali numero di lotto, partita o serie, fonti e numero di articoli); tutte le disposizioni del caso che sono state soddisfatte; norme o altri documenti normativi seguiti (ad esempio norme e specifiche tecniche nazionali) indicati in modo preciso, completo e chiaro; tutte le eventuali informazioni supplementari necessarie (ad esempio classe, categoria); data di rilascio della dichiarazione; firma e titolo o marchio equivalente della persona autorizzata; dichiarazione secondo la quale la dichiarazione di conformità viene rilasciata sotto la totale responsabilità del fabbricante ed eventualmente del suo rappresentante autorizzato. nome, l’indirizzo e il numero di identificazione dell’organismo notificato (se interviene nella procedura di valutazione della conformità) nome e indirizzo della persona che conserva la documentazione tecnica.
; tutte le disposizioni del caso che sono state soddisfatte; norme o altri documenti normativi seguiti (ad esempio norme e specifiche tecniche nazionali) indicati in modo preciso, completo e chiaro; tutte le eventuali informazioni supplementari necessarie (ad esempio classe, categoria); data di rilascio della dichiarazione; firma e titolo o marchio equivalente della persona autorizzata; dichiarazione secondo la quale la dichiarazione di conformità viene rilasciata sotto la totale. responsabilità del fabbricante ed eventualmente del suo rappresentante autorizzato. nome, l’indirizzo e il numero di identificazione dell’organismo notificato (se interviene nella procedura di valutazione della conformità) nome e indirizzo della persona che conserva la documentazione tecnica.")
70
GLI ORGANISMI NOTIFICATI
Gli Organismi Notificati sono enti pubblici o privati autorizzati a svolgere attività di certificazione, nell’ambito di una specifica direttiva, dalle Autorità Competenti dei paesi in cui hanno sede. Gli Organismi vengono notificati da ogni Stato Membro alla Commissione Europea, la quale assegna loro un numero identificativo, inserendolo in un apposito elenco. IN ITALIA GLI ORGANISMI NOTIFICATI SONO: CERTIFICATO CE: Documento con l’ Organismo Notificato certifica di aver svolto un processo di valutazione della rispondenza di un dispositivo medico alle disposizioni applicabili della direttiva di riferimento.

71
OBBLIGHI DELL’ORGANISMO NOTIFICATO
disporre di personale ed apparecchiature adeguate per espletare le procedure di valutazione di conformità. In particolare il personale scientifico in organico dovrà essere in numero sufficiente e competente per valutare, anche sul piano medico, la funzionalità e le prestazioni dei dispositivi; possedere i requisiti di indipendenza, imparzialità, riservatezza, massima integrità professionale e massima competenza; dotarsi di un manuale di qualità; assicurare la copertura economica di bilancio ed assicurativa; dimostrare che i locali interessati, ed i rispettivi impianti, garantiscono le norme di igiene ambientale e sicurezza del lavoro; accertarsi che anche le strutture eventualmente utilizzate, diverse da quelle dell’Organismo, posseggano i requisiti previsti.

72
ITER VALUTAZIONE CONFORMITÀ
La valutazione di conformità di un prodotto ai requisiti essenziali previsti dalla direttiva, viene eseguita secondo degli specifici moduli che tengono conto sia degli aspetti di progettazione che di fabbricazione. I moduli di certificazione rappresentano un precisa procedura nella quale vengono identificati obblighi per il fabbricante ed eventualmente per l’Organismo Notificato.

73
CLASSE DISPOSITIVOITER DI VALUTAZIONE
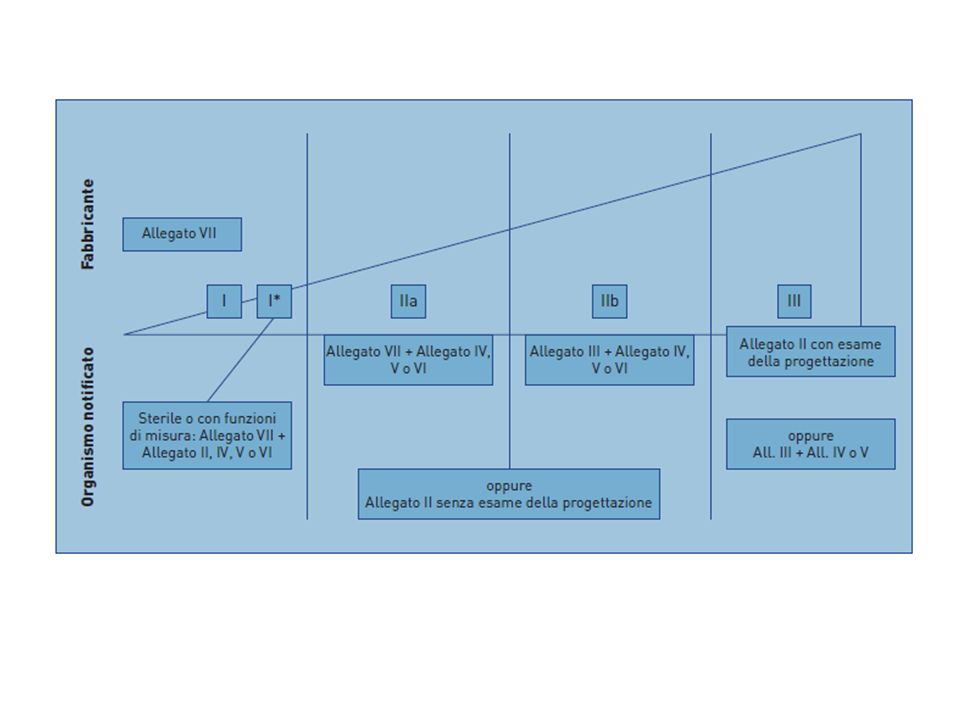
A seconda della CLASSE del DISPOSITVO il Fabbricante è tenuto a seguire un inter diverso di valutazione di conformità. Gli iter sono descritti negli ALLEGATI della DIRETTIVA. Ad ogni allegato corrisponde un MODULO Allegato II = modulo H Allegato III = modulo B Allegato IV = modulo F Allegato V = modulo D Allegato VI = modulo E Allegato VII = modulo A.

74
CLASSE ALLEGATI E PROCEDURA
CLASSE DM ALLEGATI E PROCEDURA Classe I allegato VII (Dichiarazione di Conformità CE) Classe Is e Im allegato VII + allegato IV (Verifica CE) oppure allegato VII + allegato V (Garanzia di qualità della produzione) oppure allegato VII + allegato VI (Garanzia di qualità del prodotto) oppure allegato II (Sistema completo di garanzia di qualità, sezione 3 - non si applica la sezione 4 esame della progettazione) Classe II a allegato VII + allegato IV oppure allegato VII + allegato V oppure allegato VII + allegato VI oppure allegato II (Sistema completo di garanzia di qualità, sezione 3 - non si applica la sezione 4, esame della progettazione) Classe II b allegato II (sezione 3, non si applica la sezione 4) oppure allegato III (Certificazione CE di tipo) + allegato IV oppure allegato III + allegato V oppure allegato III + allegato VI Classe III allegato II (applicazione sia della sezione 3 che della sezione 4) oppure allegato III + allegato IV oppure allegato III + allegato V
 Classe Is e Im. allegato VII + allegato IV (Verifica CE) oppure. allegato VII + allegato V (Garanzia di qualità della produzione) oppure. allegato VII + allegato VI (Garanzia di qualità del prodotto) oppure. allegato II (Sistema completo di garanzia di qualità, sezione 3 - non si applica la sezione 4 esame della progettazione) Classe II a. allegato VII + allegato IV oppure. allegato VII + allegato V oppure. allegato VII + allegato VI oppure. allegato II (Sistema completo di garanzia di qualità, sezione 3 - non si applica la sezione 4, esame della progettazione) Classe II b. allegato II (sezione 3, non si applica la sezione 4) oppure. allegato III (Certificazione CE di tipo) + allegato IV oppure. allegato III + allegato V oppure. allegato III + allegato VI. Classe III. allegato II (applicazione sia della sezione 3 che della sezione 4) oppure. allegato III + allegato IV oppure. allegato III + allegato V.")
75
ALLEGATO II: Dichiarazione CE di conformità (1) (sistema completo di garanzia della qualità)
IL FABBRICANTE DEVE Garantire la conformità dei prodotti alle disposizioni della direttiva in tutte le fasi dalla progettazione alla produzione. Redigere una documentazione aggiornata sistematicamente ed ordinata sotto forma di procedure scritte. Presentare una domanda di valutazione del proprio sistema di qualità, applicato ad un dispositivo (o ad una singola tipologia di dispositivi), all’Organismo Notificato . Redige una dichiarazione di conformità che riguarda uno o più dispositivi chiaramente identificati con il nome del prodotto, il relativo codice o altro riferimento non ambiguo. Presentare domanda di esame del fascicolo di progettazione del dispositivo da certificare.
, all’Organismo Notificato . Redige una dichiarazione di conformità che riguarda uno o più dispositivi chiaramente identificati con il nome del prodotto, il relativo codice o altro riferimento non ambiguo. Presentare domanda di esame del fascicolo di progettazione del dispositivo da certificare.")
76
L’ORGANISMO NOTIFICATO DEVE
ALLEGATO II: Dichiarazione CE di conformità (2) (sistema completo di garanzia della qualità) L’ORGANISMO NOTIFICATO DEVE Valutare l’idoneità del sistema anche a seguito di ispezione ai siti produttivi. Rilasciare un certificato CE di approvazione del sistema completo di garanzia della qualità. Effettuare ispezioni periodiche o impreviste di sorveglianza presso il fabbricante. Rilascia al fabbricante un certificato CE di esame della progettazione. PARTICOLARITÀ Per i dispositivi di classe IIa l’Organismo Notificato valuta anche la conformità della documentazione tecnica per almeno un esemplare rappresentativo per ciascuna sottocategoria di prodotti (sottocategoria:serie di dispositivi con settori di utilizzo comuni o tecnologie comuni). Per i dispositivi di classe IIb l’Organismo Notificato valuta anche la conformità della documentazione tecnica per almeno un esemplare rappresentativo per ciascun gruppo generico di dispositivi (gruppo generico: serie di dispositivi per i quali è previsto un identico ed analogo utilizzo e che condividono la stessa tecnologia, cosicché possono essere classificati in modo generico, senza tenere conto di caratteristiche specifiche)
 (sistema completo di garanzia della qualità) L’ORGANISMO NOTIFICATO DEVE. Valutare l’idoneità del sistema anche a seguito di ispezione ai siti produttivi. Rilasciare un certificato CE di approvazione del sistema completo di garanzia della qualità. Effettuare ispezioni periodiche o impreviste di sorveglianza presso il fabbricante. Rilascia al fabbricante un certificato CE di esame della progettazione. PARTICOLARITÀ. Per i dispositivi di classe IIa l’Organismo Notificato valuta anche la conformità della documentazione. tecnica per almeno un esemplare rappresentativo per ciascuna sottocategoria di prodotti (sottocategoria:serie di dispositivi con settori di utilizzo comuni o tecnologie comuni). Per i dispositivi di classe IIb l’Organismo Notificato valuta anche la conformità della documentazione tecnica per almeno un esemplare. rappresentativo per ciascun gruppo generico di dispositivi (gruppo generico: serie di dispositivi. per i quali è previsto un identico ed analogo utilizzo e che condividono la stessa tecnologia, cosicché. possono essere classificati in modo generico, senza tenere conto di caratteristiche specifiche)")
77
L’ORGANISMO NOTIFICATO DEVE
ALLEGATO III: Certificazione del Dispositivo (relativo alla progettazione) IL FABBRICANTE DEVE Presentare all’Organismo Notificato una domanda di esame di un esemplare-tipo rappresentativo della produzione di un dispositivo, allegando documentazione tecnica (schemi di progettazione,metodi di fabbricazione previsti, norme applicate, analisi dei rischi, prove effettuate, dati clinici di supportoecc.). Fornire, inoltre, all’Organismo Notificato uno o più esemplari del dispositivo tipo. L’ORGANISMO NOTIFICATO DEVE Esaminare e valutare la documentazione Svolge o far svolgere i controlli e prove sull’esemplare tipo messo a disposizione per verificarne la conformità alle disposizioni della direttiva. Rilasciare al fabbricante un certificato CE di esame della progettazione dell’esemplare tipo.
 IL FABBRICANTE DEVE. Presentare all’Organismo Notificato una domanda di esame di un esemplare-tipo rappresentativo della produzione di un dispositivo, allegando documentazione tecnica (schemi di progettazione,metodi di fabbricazione previsti, norme applicate, analisi dei rischi, prove effettuate, dati clinici di supportoecc.). Fornire, inoltre, all’Organismo Notificato uno o più esemplari del dispositivo tipo. L’ORGANISMO NOTIFICATO DEVE. Esaminare e valutare la documentazione. Svolge o far svolgere i controlli e prove sull’esemplare tipo messo a disposizione per verificarne la conformità alle disposizioni della direttiva. Rilasciare al fabbricante un certificato CE di esame della progettazione dell’esemplare tipo.")
78
L’ORGANISMO NOTIFICATO DEVE
ALLEGATO IV: Verifica CE (relativo alla produzione di un lotto) IL FABBRICANTE DEVE Presentare all’Organismo Notificato una domanda di esame di una produzione di un lotto di un dispositivo, allegando documentazione atta a dimostrare la omogeneità della produzione e le prove ed i controlli effettuati. Fornire all’Organismo anche un campione significativo del lotto. L’ORGANISMO NOTIFICATO DEVE Accertarsi dall’analisi della documentazione che il lotto sia omogeneo. Svolgere o fa svolgere i controlli sui campioni per verificare la rispondenza del lotto all’esemplare tipo progettato. Se le prove soddisfano le disposizioni previste dalla normativa, rilascia un certificato di Verifica CE della produzione del lotto. IL FABBRICANTE predispone una dichiarazione di conformità indicando il lotto oggetto di certificazione
 IL FABBRICANTE DEVE. Presentare all’Organismo Notificato una domanda di esame di una produzione di un lotto di un dispositivo, allegando documentazione atta a dimostrare la omogeneità della produzione e le prove ed i controlli effettuati. Fornire all’Organismo anche un campione significativo del lotto. L’ORGANISMO NOTIFICATO DEVE. Accertarsi dall’analisi della documentazione che il lotto sia omogeneo. Svolgere o fa svolgere i controlli sui campioni per verificare la rispondenza del lotto all’esemplare tipo progettato. Se le prove soddisfano le disposizioni previste dalla normativa, rilascia un certificato di Verifica CE della produzione del lotto. IL FABBRICANTE predispone una dichiarazione di conformità indicando il lotto oggetto di certificazione.")
79
L’ORGANISMO NOTIFICATO DEVE
ALLEGATO V: Dichiarazione CE di Conformità (relativo solo alla Garanzia di qualità della produzione di un dispositivo) IL FABBRICANTE DEVE Presentare una domanda di valutazione del proprio sistema di qualità applicato alla produzione di un dispositivo (o ad una singola tipologia di dispositivi) all’Organismo Notificato. Tutte le disposizioni del fabbricante devono figurare in una documentazione aggiornata sistematicamente ed ordinata sotto forma di procedure scritte. Effettuare ispezioni periodiche o impreviste di sorveglianza presso il fabbricante L’ORGANISMO NOTIFICATO DEVE Valutare l’idoneità del sistema anche a seguito di ispezione ai siti produttivi Rilascia al fabbricante il certificato di approvazione del sistema di qualità applicato alla produzione. Per i dispositivi di classe Iia valutare anche la conformità della documentazione tecnica per almeno un esemplare rappresentativo per ciascuna sottocategoria di prodotti. IL FABBRICANTE redige una dichiarazione di conformità che riguarda uno o più dispositivi chiaramente identificati con il nome del prodotto, il relativo codice o altro riferimento non ambiguo.
 IL FABBRICANTE DEVE. Presentare una domanda di valutazione del proprio sistema di qualità applicato alla produzione di un dispositivo (o ad una singola tipologia di dispositivi) all’Organismo Notificato. Tutte le disposizioni del fabbricante devono figurare in una documentazione aggiornata sistematicamente ed ordinata sotto forma di procedure scritte. Effettuare ispezioni periodiche o impreviste di sorveglianza presso il fabbricante. L’ORGANISMO NOTIFICATO DEVE. Valutare l’idoneità del sistema anche a seguito di ispezione ai siti produttivi. Rilascia al fabbricante il certificato di approvazione del sistema di qualità applicato alla produzione. Per i dispositivi di classe Iia valutare anche la conformità della documentazione tecnica per almeno un esemplare rappresentativo per ciascuna sottocategoria di prodotti. IL FABBRICANTE redige una dichiarazione di conformità che riguarda uno o più dispositivi chiaramente identificati con il nome del prodotto, il relativo codice o altro riferimento non ambiguo.")
80
L’ORGANISMO NOTIFICATO DEVE
ALLEGATO VI: Dichiarazione di Conformità CE (relativo alla fase di controllo finale a garanzia della qualità del prodotto) IL FABBRICANTE DEVE Procedere all’esame di ogni prodotto o di campioni rappresentativi del lotto e allo svolgimento delle prove necessarie per verificarne la conformità. Tutte le disposizioni del fabbricante per garantire il Sistema di Qualità devono figurare in una documentazione aggiornata sistematicamente ed ordinata sottoforma di procedure scritte. Presentare una domanda di valutazione del sistema di qualità applicato ai controlli finali di un dispositivo all’Organismo Notificato. L’ORGANISMO NOTIFICATO DEVE Valutare l’idoneità del sistema, anche a seguito di ispezione ai siti di prova e di conservazione, rilascia il certificato di garanzia di qualità del prodotto al fabbricante. Effettuare ispezioni periodiche o impreviste di sorveglianza presso il fabbricante. Per i dispositivi di classe IIa l’organismo Notificato valuta anche la conformità della documentazione tecnica per almeno un esemplare rappresentativo per ciascuna sottocategoria di prodotti. IL FABBRICANTE redige una dichiarazione di conformità che riguarda uno o più dispositivi chiaramenteidentificati con il nome del prodotto, il relativo codice o altro riferimento non ambiguo.
 IL FABBRICANTE DEVE. Procedere all’esame di ogni prodotto o di campioni rappresentativi del lotto e allo svolgimento delle prove necessarie per verificarne la conformità. Tutte le disposizioni del fabbricante per garantire il Sistema di Qualità devono figurare in una documentazione aggiornata sistematicamente ed ordinata sottoforma di procedure scritte. Presentare una domanda di valutazione del sistema di qualità applicato ai controlli finali di un dispositivo all’Organismo Notificato. L’ORGANISMO NOTIFICATO DEVE. Valutare l’idoneità del sistema, anche a seguito di ispezione ai siti di prova e di conservazione, rilascia il certificato di garanzia di qualità del prodotto al fabbricante. Effettuare ispezioni periodiche o impreviste di sorveglianza presso il fabbricante. Per i dispositivi di classe IIa l’organismo Notificato valuta anche la conformità della documentazione tecnica per almeno un esemplare rappresentativo per ciascuna sottocategoria di prodotti. IL FABBRICANTE redige una dichiarazione di conformità che riguarda uno o più dispositivi chiaramenteidentificati con il nome del prodotto, il relativo codice o altro riferimento non ambiguo.")
81
ALLEGATO VII: Dichiarazione di Conformità CE (relativo a progettazione e produzione di un dispositivo) IL FABBRICANTE Garantisce (tramite la dichiarazione di conformità) che i prodotti soddisfano le disposizioni applicabili della direttiva e predispone la documentazione tecnica (Fascicolo Tecnico), relativa ai dispositivi progettati e prodotti, che viene tenuta a disposizione dell’Autorità competente. Il fascicolo tecnico deve consentire di valutare la conformità del prodotto ai requisiti della direttiva e deve contenere documentazione specifica su progettazione e fabbricazione nonché documentazione su sicurezza ed efficacia. Questo è l’unico allegato che non prevede l’intervento di un Organismo Notificato.
 che i prodotti soddisfano le disposizioni. applicabili della direttiva e predispone la documentazione tecnica (Fascicolo Tecnico), relativa ai dispositivi progettati e prodotti, che viene tenuta a disposizione dell’Autorità competente. Il fascicolo tecnico deve consentire di valutare la conformità del prodotto ai requisiti della direttiva e deve contenere documentazione specifica su progettazione e fabbricazione nonché documentazione su sicurezza ed efficacia. Questo è l’unico allegato che non prevede l’intervento di un Organismo Notificato.")
82
PROCEDURE COMPLETE DI VALUTAZIONE
Gli allegati andranno eventualmente combinati tra loro al fine di definire procedure complete di valutazione (progettazione + produzione). Il tipo di intervento dell’Organismo Notificato sarà graduale e “dosato” in relazione alla classe del dispositivo: Classe III e IIb: la valutazione dell’Organismo riguarda tutti gli aspetti connessi al dispositivo medico che vanno dalla progettazione alla produzione. Classe IIa: la valutazione dell’Organismo riguarda, solo gli aspetti della produzione. Classe Is e Im: la valutazione dell’Organismo riguarda solo alcuni aspetti della produzione (quelli legati alla sterilizzazione per i dispositivi di classe Is, quelli legati agli aspetti metrologici per i dispositivi di classe Im). Classe I: non è previsto l’intervento dell’Organismo; progettazione e produzione vengono valutate ed accertate solo dal fabbricante
. Il tipo di intervento dell’Organismo Notificato sarà graduale e dosato in relazione alla classe del dispositivo: Classe III e IIb: la valutazione dell’Organismo riguarda tutti gli aspetti connessi al dispositivo medico che vanno dalla progettazione alla produzione. Classe IIa: la valutazione dell’Organismo riguarda, solo gli aspetti della produzione. Classe Is e Im: la valutazione dell’Organismo riguarda solo alcuni aspetti della produzione (quelli legati alla sterilizzazione per i dispositivi di classe Is, quelli legati agli aspetti metrologici per i dispositivi di classe Im). Classe I: non è previsto l’intervento dell’Organismo; progettazione e produzione vengono valutate ed accertate solo dal fabbricante.")
84
Nella direttiva sui DM (settore dove l’avanzamento tecnologico è continuo e rapido) sono riportate le specifiche tecniche? ?

85
? Come fa il Produttore a Verificare la Rispondenza ai Requisiti Essenziali?

86
LE NORME TECNICHE ARMONIZZATE
Rappresentano lo stato dell’arte nel settore, il loro rispetto assicura una presunzione di conformità che il fabbricante dovrà dimostrare in caso di non utilizzo delle norme stesse.

87
LE NORME TECNICHE ARMONIZZATE
NORMA TECNICA: “specificazione tecnica approvata da un organismo riconosciuto a svolgere attività normativa per applicazione ripetuta o continua la cui osservazione non è obbligatoria ed appartiene ad una delle seguenti categorie” Norma Internazionale (elaborata dall’ISO o, per il settore elettrico da IEC) Norma Europea (elaborata da CEN o CENELEC) Norma Nazionale (elaborata, in Italia, da UNI o CEI) Il termine ‘Armonizzate’ indica che sono adottate a livello Europeo, su mandato della Commissione, dai Comitati Europei di Normalizzazione. Queste norme sono: Consensuali: devono essere approvate con il consenso di coloro che hanno partecipato ai lavori; Volontarie:le parti interessate si impongono spontaneamente di rispettarle; Democratiche, secondo cui tutte le parti economico-sociali interessate possono partecipare ai lavori di compilazione.
 Norma Europea (elaborata da CEN o CENELEC) Norma Nazionale (elaborata, in Italia, da UNI o CEI) Il termine ‘Armonizzate’ indica che sono adottate a livello Europeo, su mandato della Commissione, dai Comitati Europei di Normalizzazione. Queste norme sono: Consensuali: devono essere approvate con il consenso di coloro che hanno partecipato ai lavori; Volontarie:le parti interessate si impongono spontaneamente di rispettarle; Democratiche, secondo cui tutte le parti economico-sociali interessate possono partecipare ai lavori di compilazione.")
88
ALCUNE NORME TECNICHE Norma Tecnica EN 14971:
“Applicazione della Gestione del Rischio dei Dispositivi Medici” Il fabbricante, come previsto dalla norma, deve emettere giudizi relativi alla sicurezza di un dispositivo medico, tenendo conto dello stato dell’arte ed attuando il processo di Gestione dei Rischi che deve includere le seguenti parti: Analisi dei Rischi Valutazione dei Rischi Controllo dei Rischi Valutazione del rischio residuo Informazioni di produzione e post produzione Norma Tecnica EN : “Dispositivi Medici: Sistema di Gestione della Qualità” I punti chiavi sui quali la norma si basa sono l’analisi dei rischi, sviluppata con approccio di identificazione, valutazione, prevenzione e valutazione del rischio residuo, comunicazione e informazione verso l’utilizzatore per una gestione del dispositivo in un’ottica di certa rintracciabilità del prodotto e delle sue componenti critiche, sistema di gestione aziendale e controllo di processo

89
Se il fabbricante non rispetta la direttiva…a cosa va incontro?

91
Decreto lgs. 24 febbraio 1997, n. 46 emendato col D. lgs. 25.01.2010,
n.37 - Recepimento Direttiva 2007/47/CE

92
LA CODIFICA NAZIONALE DEI DISPOSITIVI MEDICI
(CND)
")
93
CODIFICHE: Codifica CND (1)
La Commissione Unica sui Dispositivi Medici: Art. 57 della Legge n. 289 del 27/12/2002 (Finanziaria 2003): istituisce la Commissione Unica sui Dispositivi Medici (CUD), organo Tecnico-consultivo del Ministero della Salute. I compiti della CUD (Art. 57 L.289/2002): Valuta le tecnologie avanzate e effettua attività di “Technology assessment” Definisce e aggiorna il Repertorio dei dispositivi medici Classifica i prodotti in classi e sottoclassi specifiche Indica il prezzo di riferimento (per classe) D.M. del 1/10/2003: Nomina dei componenti individuati tra i maggiori esperti del settore Composizione CUD: Presidente : il Ministro della Salute o un vice presidente da lui designato 5 componenti nominati dal Ministro della Salute 1 componente nominato dal Ministero dell’Economia e delle Finanze 7 componenti nominati dalla Conferenza dei Presidenti delle Regioni e delle Province autonome 2 componenti di diritto (Direzione Generale dei Farmaci e dei Dispositivi medici, Istituto Superiore di Sanità)
: istituisce la Commissione Unica sui Dispositivi Medici (CUD), organo Tecnico-consultivo del Ministero della Salute. I compiti della CUD (Art. 57 L.289/2002): Valuta le tecnologie avanzate e effettua attività di Technology assessment Definisce e aggiorna il Repertorio dei dispositivi medici. Classifica i prodotti in classi e sottoclassi specifiche. Indica il prezzo di riferimento (per classe) D.M. del 1/10/2003: Nomina dei componenti individuati tra i maggiori esperti del settore. Composizione CUD: Presidente : il Ministro della Salute o un vice presidente da lui designato. 5 componenti nominati dal Ministro della Salute. 1 componente nominato dal Ministero dell’Economia e delle Finanze. 7 componenti nominati dalla Conferenza dei Presidenti delle Regioni e delle Province autonome. 2 componenti di diritto (Direzione Generale dei Farmaci e dei Dispositivi medici, Istituto Superiore di Sanità)")
94
Codifica CND (2) D.M. 22/09/2005 (Pubblicato sulla G.U. n. 286 del 9/12/2005) Art. 1 “E’ approvata la classificazione dei dispositivi medici elaborata dalla CUD ai sensi dell’art. 57 della Legge 27 Dicembre 2002 n. 289.” 2. La classificazione di cui al comma 1, riferita ai dispositivi medici disciplinati dai decreti legislativi 14 dicembre 1992, n. 507 (impiantabili attivi) e 24 febbraio 1997, n. 46 e successive modificazioni, costituisce la prima parte della “Classificazione nazionale dei dispositivi medici” (CND), destinata ad essere utilizzata in tutte le attività attinenti alla commercializzazione dei dispositivi sul territorio nazionale e alle attività di sorveglianza, vigilanza e certificazione da parte delle autorità competenti e degli organismi notificati. La seconda parte della classificazione, riferita ai dispositivi medico-diagnostici in vitro, sarà approvata con successivo provvedimento previa deliberazione della Commissione unica sui dispositivi medici.
 e 24 febbraio 1997, n. 46 e successive modificazioni, costituisce la prima parte della Classificazione nazionale dei dispositivi medici (CND), destinata ad essere utilizzata in tutte le attività attinenti alla commercializzazione dei dispositivi sul territorio nazionale e alle attività di sorveglianza, vigilanza e certificazione da parte delle autorità competenti e degli organismi notificati. La seconda parte della classificazione, riferita ai dispositivi medico-diagnostici in vitro, sarà approvata con successivo provvedimento previa deliberazione della Commissione unica sui dispositivi medici.")
95
Codifica CND (3) La necessità di disporre di tale tipo di classificazione deriva dal fatto che gli altri sistemi di classificazione esistenti e utilizzati in Europa e nel mondo, pur comprendendo la maggioranza dei dispositivi presenti sul mercato, non permettono di raggruppare i dispositivi in categorie omogenee di prodotti e cioè in categorie di dispositivi destinati ad effettuare un intervento diagnostico o terapeutico simile. Il poter disporre di un tale tipo di classificazione offrirà dei vantaggi notevoli quale quello di poter scambiare informazioni, con un linguaggio comune, tra tutti i soggetti che si occupano o gestiscono il settore dei dispositivi medici. L’obiettivo è consentire: di monitorare in maniera più efficace sia il consumo che l’uso dei dispositivi permettere una migliore valutazione degli incidenti comparativamente per singole tipologie nell’ambito della vigilanza facilitere e rendere più trasparenti i processi d’acquisto da parte del Sistema sanitario nazionale in quanto permetterà la definizione di prezzi di riferimento per classi e sottoclassi omogenee

96
Codifica CND (4) La classificazione CND
La CND classifica in classi omogenee tutti i dispositivi medici in commercio in Italia definiti tali dall’art.1 del D.Lgs. n. 507 del 14/12/1992 (Direttiva 90/385/CEE) e dall’art 1 del D.Lgs. n. 46 del 24/12/1997 (Direttiva 93/42/CEE). Non sono presenti nella CND: Medicinali (D.Lgs. 178/91) Prodotti cosmetici (D.Lgs. 713/86) Sangue umano e suoi derivati Organi, tessuti o cellule di origine umana e prodotti comprendenti o derivati da tessuti o cellule di origine umana Organi, tessuti o cellule di origine animale, salvo che il dispositivo non sia fabbricato utilizzando tessuto animale reso non vitale o prodotti non vitali derivati da tessuto animale Dispositivi di protezione individuale (D.Lgs. 475/92) Modalità di aggiornamento della CND Le revisioni sono prodotte con cadenza almeno annuale e previa consultazione, a livello tecnico, delle Regioni e delle Associazioni industriali. “classi omogenee”: gruppo di dispositivi medici che, pur potendosi differenziare per caratteristiche tecnologiche, in rapporto alla loro applicazione o metodica clinica di utilizzo, permettono un intervento terapeutico o diagnostico simile.
 e dall’art 1 del D.Lgs. n. 46 del 24/12/1997 (Direttiva 93/42/CEE). Non sono presenti nella CND: Medicinali (D.Lgs. 178/91) Prodotti cosmetici (D.Lgs. 713/86) Sangue umano e suoi derivati. Organi, tessuti o cellule di origine umana e prodotti comprendenti o derivati da tessuti o cellule di origine umana. Organi, tessuti o cellule di origine animale, salvo che il dispositivo non sia fabbricato utilizzando tessuto animale reso non vitale o prodotti non vitali derivati da tessuto animale. Dispositivi di protezione individuale (D.Lgs. 475/92) Modalità di aggiornamento della CND. Le revisioni sono prodotte con cadenza almeno annuale e previa consultazione, a livello tecnico, delle Regioni e delle Associazioni industriali. classi omogenee : gruppo di dispositivi medici che, pur potendosi differenziare per caratteristiche tecnologiche, in rapporto alla loro applicazione o metodica clinica di utilizzo, permettono un intervento terapeutico o diagnostico simile.")
97
Codifica CND (5) ALBERO GERARCHICO UNIVOCO A 7 LIVELLI
La classificazione CND Codifica alfa-numerica; Differenziazione dei prodotti per metodica clinica e/o distretto anatomico di utilizzo; Struttura ad albero gerarchico multilivello; Aggregazione dei dispositivi medici in Categorie, Gruppi e Tipologie. Gruppo Tip.1 Tip.2 Tip.3 Tip.4 Tip.5 Categoria ALBERO GERARCHICO UNIVOCO A 7 LIVELLI 22 Categorie anatomico-funzionali 145 Gruppi 719 Tipologia 1 1659 Tipologia 2 2249 Tipologia 3 1574 Tipologia 4 234 Tipologia 5 Al 12/02/2010

98
Codifica CND (6) La Categoria
Costituisce la 1° stratificazione gerarchica. Sono presenti 22 categorie anatomico/funzionali contraddistinte da una lettera dell'alfabeto. Le categorie hanno come criterio di classificazione quello di contenere, ciascuna, dispositivi utilizzati su uno stesso specifico apparato, distretto o organo anatomico o in sostituzione di essi, oppure dispositivi caratterizzati da una affinità di utilizzo, destinazione d'uso o di metodica clinica oppure dispositivi che sono regolamentati da una specifica direttiva europea diversa dalla 93/42/CE o che sono gestiti in modo particolare dalle ASL/Aziende ospedaliere o che seguono delle regole specifiche per la prescrizione o il rimborso. 8 CATEGORIE ANATOMICHE (per distretto anatomico di utilizzo) B DISPOSITIVI EMOTRASFUSIONE ED EMATOLOGIA C DISPOSITIVI PER APPARATOCARDIOCIRCOLATORIO F DISPOSITIVI PER DIALISI, EMO ED EMODIAFILTRAZIONE G DISPOSITIVI PER APPARATO GASTROINTESTINALE N DISPOSITIVI PER IL SISTEMA NERVOSO E MIDOLLARE Q DISPOSITIVI PER ODONTOIATRIA, OFTALMOLOGIA E OTORINOLARINGOIATRIA R DISPOSITIVI PER APPARATO RESPIRATORIO E ANESTESIA U DISPOSITIVI PER APPARATO UROGENITALE 9 CATEGORIE FUNZIONALI (per metodica clinica di utilizzo) A DISPOSITIVI DA SOMMINISTRAZIONE, PRELIEVO E RACCOLTA D DISINFETTANTI, ANTISETTICI E PROTEOLITICI (D.Lgs.46/97) H DISPOSITIVI DA SUTURA K DISPOSITIVI CHIRUR. MINI-INVASIVA ED ELETTROCHIRURGIA L STRUMENTARIO CHIRURGICO PLURIUSO M DISPOSITIVI PER MEDICAZIONE GENERALI E SPECIALI S PRODOTTI PER STERILIZZAZIONE T DISPOSITIVI MEDICI DI PROTEZIONE E AUSILI PER INCONTINENZA (D.Lgs.46/97) V DISPOSITIVI VARI 5 CATEGORIE SPECIALI (per criteri specifici) J DISPOSITIVI IMPIANTABILI ATTIVI P DISPOSITIVI PROTESICI E PRODOTTI PER OSTEOSINTESI Y SUPPORTI O AUSILI TECNICI PER DISABILI W DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO (D. Lgs. 332/2000) Z APPARECCHIATURE SANITARIE
 B DISPOSITIVI EMOTRASFUSIONE ED EMATOLOGIA C DISPOSITIVI PER APPARATOCARDIOCIRCOLATORIO F DISPOSITIVI PER DIALISI, EMO ED EMODIAFILTRAZIONE G DISPOSITIVI PER APPARATO GASTROINTESTINALE N DISPOSITIVI PER IL SISTEMA NERVOSO E MIDOLLARE Q DISPOSITIVI PER ODONTOIATRIA, OFTALMOLOGIA E OTORINOLARINGOIATRIA R DISPOSITIVI PER APPARATO RESPIRATORIO E ANESTESIA U DISPOSITIVI PER APPARATO UROGENITALE. 9 CATEGORIE FUNZIONALI (per metodica clinica di utilizzo) A DISPOSITIVI DA SOMMINISTRAZIONE, PRELIEVO E RACCOLTA D DISINFETTANTI, ANTISETTICI E PROTEOLITICI (D.Lgs.46/97) H DISPOSITIVI DA SUTURA K DISPOSITIVI CHIRUR. MINI-INVASIVA ED ELETTROCHIRURGIA L STRUMENTARIO CHIRURGICO PLURIUSO M DISPOSITIVI PER MEDICAZIONE GENERALI E SPECIALI S PRODOTTI PER STERILIZZAZIONE T DISPOSITIVI MEDICI DI PROTEZIONE E AUSILI PER INCONTINENZA (D.Lgs.46/97) V DISPOSITIVI VARI. 5 CATEGORIE SPECIALI (per criteri specifici) J DISPOSITIVI IMPIANTABILI ATTIVI P DISPOSITIVI PROTESICI E PRODOTTI PER OSTEOSINTESI Y SUPPORTI O AUSILI TECNICI PER DISABILI W DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO (D. Lgs. 332/2000) Z APPARECCHIATURE SANITARIE.")
99
Codifica CND (7) Il Gruppo La Tipologia
Costituisce la 2° stratificazione gerarchica Sono presenti 145 gruppi anatomico/funzionali di dispositivi medici che rappresentano le varie differenziazioni in cui si distinguono i dispositivi contenuti nelle categorie. Vengono contraddistinti da un numero a due cifre da 01 a 99 per ognuna delle categorie Il numero 90 individua i gruppi contenenti dispositivi con caratteristiche varie, non riconducibili ai gruppi già esistenti. Il numero 99 "Altri", viene riservato a dispositivi non compresi nei gruppi già esistenti, da classificare nei successivi aggiornamenti. Il codice riservato al termine generico "Altri" deve essere utilizzato dagli utenti esclusivamente nei casi in cui il dispositivo medico non sia collocabile nei gruppi già esistenti e sarà oggetto di classificazione nei successivi aggiornamenti. La Tipologia Rappresenta la 3° stratificazione gerarchica Se del caso, si espande in più sottolivelli di dettaglio (1°, 2°, 3°, 4° e 5°) Nell’ambito del Gruppo di appartenenza ogni Tipologia contiene dispositivi caratterizzati da una ancor maggior affinità di utilizzo, destinazione d'uso o di metodica clinica In caso di dubbio, per una corretta collocazione o ricerca, si dovranno considerare sempre le caratteristiche peculiari del dispositivo medico preso in esame (cioè le caratteristiche anatomico-funzionali e di destinazione d'uso attribuite dal fabbricante) Come già detto per i gruppi, la suddivisione "Altri", con il numero 99 nel 1° sottolivello di dettaglio, viene riservata a dispositivi non compresi nelle tipologie già esistenti, da classificare nei successivi aggiornamenti Il codice riservato al termine generico "Altri" deve essere utilizzato dagli utenti esclusivamente nei casi in cui il dispositivo medico non sia collocabile nelle suddivisioni già esistenti e tale tipologia sarà oggetto di continua e successiva verifica
 Nell’ambito del Gruppo di appartenenza ogni Tipologia contiene dispositivi caratterizzati da una ancor maggior affinità di utilizzo, destinazione d uso o di metodica clinica. In caso di dubbio, per una corretta collocazione o ricerca, si dovranno considerare sempre le caratteristiche peculiari del dispositivo medico preso in esame (cioè le caratteristiche anatomico-funzionali e di destinazione d uso attribuite dal fabbricante) Come già detto per i gruppi, la suddivisione Altri , con il numero 99 nel 1° sottolivello di dettaglio, viene riservata a dispositivi non compresi nelle tipologie già esistenti, da classificare nei successivi aggiornamenti. Il codice riservato al termine generico Altri deve essere utilizzato dagli utenti esclusivamente nei casi in cui il dispositivo medico non sia collocabile nelle suddivisioni già esistenti e tale tipologia sarà oggetto di continua e successiva verifica.")
100
Codifica CND (8) CLASSIFICAZIONE NAZIONALE DISPOSITIVI MEDICI
1° e 2° Livello Z Apparecchiature Sanitarie Z11 Bioimmagini Radioterapia Z12 Esplor. Fun. Ter.

101
Z11020105 Codifica CND (9) Tipologia 3° liv
GCC in stazione fissa a terta multipla – con acquisizione “Total Body” Categoria App. Sanitarie Tipologia 1° liv Strumentazione per medicina nucleare Gruppo Strumentazione per bioimmagini e radioterapia Tipologia 2° liv Gamma camera computerizzata

102
Codifica CND (10) A DISPOSITIVI DA SOMMINISTRAZIONE, PRELIEVO E RACCOLTA A02 SIRINGHE A0201 SIRINGHE MONOUSO A020101 SIRINGHE A PERDITA DI RESISTENZA A020102 SIRINGHE DA INFUSIONE ED IRRIGAZIONE MONOUSO A SIRINGHE DA INFUSIONE ED IRRIGAZIONE MONOUSO CON CONO LUER A SIRINGHE DA INFUSIONE ED IRRIGAZIONE MONOUSO CON CONO LUER A DUE PEZZI A SIRINGHE DA INFUSIONE ED IRRIGAZIONE MONOUSO CON CONO LUER A DUE PEZZI CON AGO A SIRINGHE DA INFUSIONE ED IRRIGAZIONE MONOUSO CON CONO LUER A DUE PEZZI SENZA AGO

103
REPERTORIO DEI DISPOSITIVI MEDICI
Ai sensi della L. 289/2002 è istituito il repertorio dei dispositivi medici: Il Fabbricante attenendosi a quanto disposto dal 46/97 (requisiti essenziali, …) deve richiedere per l’immissione sul mercato l’assegnazione di un numero di “repertorio” compilando una scheda dettagliata delle caratteristiche del Dispositivo: schede tecniche, istruz. d’uso, ecc, del prezzo di listino che diviene ufficiale e di riferimento per gli acquisti da parte del SSN. Tra le informazioni obbligatorie è l’attribuzione della cod. CND. Da qui il collegamento tra cod. CND e Repertorio.
 deve richiedere per l’immissione sul mercato l’assegnazione di un numero di repertorio compilando una scheda dettagliata delle caratteristiche del Dispositivo: schede tecniche, istruz. d’uso, ecc, del prezzo di listino che diviene ufficiale e di riferimento per gli acquisti da parte del SSN. Tra le informazioni obbligatorie è l’attribuzione della cod. CND. Da qui il collegamento tra cod. CND e Repertorio.")
Presentazioni simili





















 Qualità. I progetti selezionati per l'erogazione di un finanziamento devono dimostrarsi di elevato.>")
