Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
In un insieme canonico, il valore medio di una qualsiasi grandezza fisica A si può ottenere dalla media su tutte le copie diverse del sistema, secondo l’espressione: Passando a variabili continue nello spazio delle fasi:

2
quando si può scrivere: si ottiene: La superficie di energia potenziale presenta ampie zone stericamente proibite, con profondi minimi Schematicamente si ha: Da qui l’importanza di una esplorazione delle zone a energia potenziale minore

3
Metodo Montecarlo Calcolo di un integrale Selezionando n numeri a caso nell’intervallo Per n grandi:

5
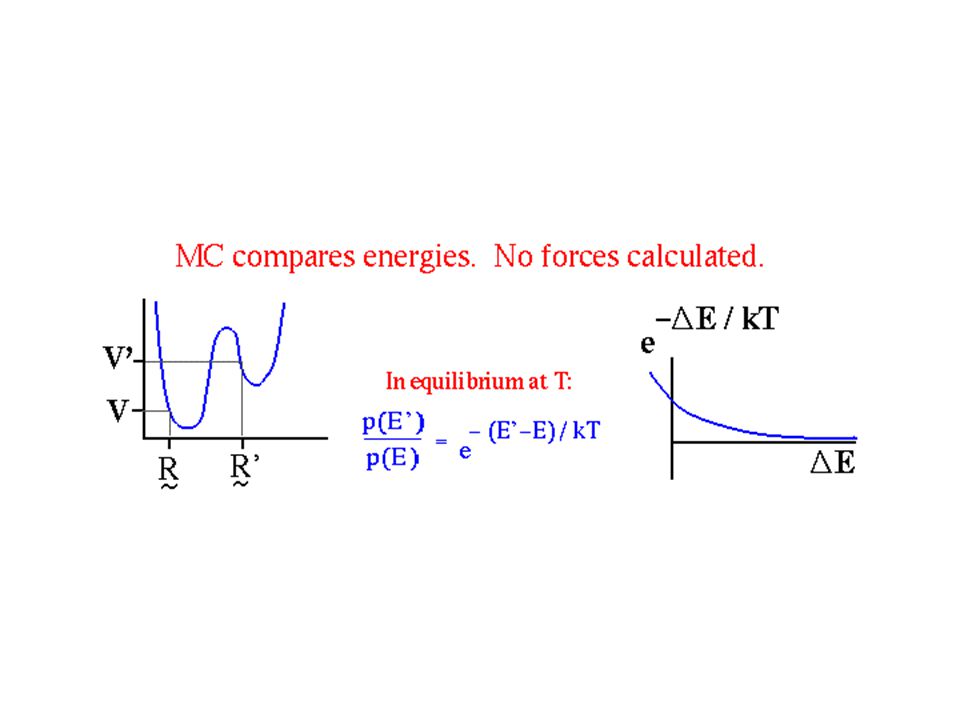
Metropolis Monte Carlo The most common technique employed in MC simulations is the Metropolis algorithm and involves the following steps: 1.Construct an initial configuration for a molecule. 2.Make a random trial change in one of the degrees of freedom (e.g. dihedral angle) in a random molecule. 3.Compute the change in energy ( E) of the system due to the trial change. 4.If,accept the new configuration. 5.If,compute the transition probability, Generate a random number, r in the interval [0,1], then accept the move, if or retain the previous configuration, if By comparing the Boltzmann weighted energy difference to a random number, a few higher energy conformers will be accepted, and energy barriers can in principle be crossed.
 in a random molecule. 3.Compute the change in energy ( E) of the system due to the trial change. 4.If,accept the new configuration. 5.If,compute the transition probability, Generate a random number, r in the interval [0,1], then accept the move, if or retain the previous configuration, if By comparing the Boltzmann weighted energy difference to a random number, a few higher energy conformers will be accepted, and energy barriers can in principle be crossed..")
6
Il rapporto tra le popolazioni è determinato dalla legge di Boltzmann: simulazione MonteCarlo Metropolis Sia V n l'energia potenziale della configurazione n-esima e V n-1 quella relativa alla configurazione precedente (n-1)-esima. Ci si trovi in una situazione in cui V n > V n-1. In tal caso la nuova configurazione viene accettata con una probabilità: e ciò viene fatto tramite la generazione di un numero casuale compreso tra 0 e 1, e se tale numero è ≤ p, la configurazione viene accettata e memorizzata, se è > p, la configurazione non viene accettata, e viene memorizzata la configurazione (n-1)-esima.
-esima..")
7
Siano date 2 configurazioni diverse r e s del sistema in studio e sia La probabilità P rs che venga generata una catena di trasformazioni che porta il sistema dalla configurazione r alla s, è a priori = alla probabilità P sr che venga generata una catena di trasformazioni che porta il sistema dalla configurazione s alla r: Distribuzione delle configurazioni numero di configurazioni r ottenute numero di configurazioni s ottenute Numero di passaggi In una simulazione infinita, il numero di configurazioni varia fino a raggiungere l’equilibrio tra i passaggi nelle 2 direzioni:

9
Poiché la distribuzione è già canonica, è sufficiente fare la media dei valori che la proprietà assume in ogni singola configurazione: dove N è il n. totale di configurazioni.

11
Molecular Dynamics Very often in Chemistry and Biochemistry, molecular structures are displayed as static, rigid entities with the exception of rotation about single bonds. However, a molecule is not a rigid, fixed struture at some minimum energy but is actually constantly vibrating at a higher energy somewhere above the relaxed potential energy surface. When this conceptual barrier is overcome, it is possible to understand that the very large structural changes taking place in biological systems can be broken down into many smaller changes. What we are in fact observing is a cumulative effect of many, small, thermally accessible changes resulting from the dynamical properties of proteins. Although these dynamical changes may not be observed experimentally, they may be calculated by theoretical methods such as Molecular Dynamics. The starting point for the Molecular Dynamics (MD) method is a well-defined microscopic description of a macroscopic physical system. This description can be a Hamiltonian or Lagrangian, or a direct expression in Newton's equations of motion. As the name suggests, the Molecular Dynamics method calculates properties using the equations of motion and allows for the calculation of both static and dynamic properties. On the other hand, the Monte Carlo method produces configurational properties.
 method is a well-defined microscopic description of a macroscopic physical system. This description can be a Hamiltonian or Lagrangian, or a direct expression in Newton s equations of motion. As the name suggests, the Molecular Dynamics method calculates properties using the equations of motion and allows for the calculation of both static and dynamic properties. On the other hand, the Monte Carlo method produces configurational properties..")
12
Molecular Dynamics simulations are sometimes referred to as deterministic in the sense that, given a set of initial coordinates and velocities with a force (F(r)) the evolution of the system over time is now determined. Also, the motion of the particles in Molecular Dynamics is collective because the collection of forces causes ALL the particles to move over time. This is distinct from the Monte Carlo method which requires external changes in the system by allowing random moves to produces the changes. The force field for a typical protein can be given as a sum of the various components including bond stretching and bending, torsional potentials, and non-bonded interactions.

13
It is then possible to calculate the forces between the atoms by taking the derivative of the potential V(r) given above and leads to the following equation Integrazione numerica delle equazioni del moto
 given above and leads to the following equation Integrazione numerica delle equazioni del moto")
14
Simulation Times, The Time Step For a large protein consisting of ~15000 atoms a Molecular Dynamics is very computationally demanding and is usually limited to 300 - 400 picoseconds of simulation time although simulation times of 1 nanosecond for a few large proteins have been performed. (1 picosecond (ps) = 1 x 10 -12 sec; 1 nanosecond (ns) = 1 x 10 -9 sec; 1000 ps = 1 ns). Molecular Dynamics is extremely useful to study properties that depend on high frequency vibrations. It is difficult to study properties such as rotations, diffusion mechanisms, and viscosity using Molecular Dynamics where local events are equilibrated on the order of 100 ps and longer. Unless small time steps are used, very significant errors in the energy will be introduced. In practice the time step is taken in femtoseconds (1 femtosecond (fs) = 1 x 10 -15 sec). To ensure numerical stability for the solution of the equations of motion, the time step must be at most 1/100 - 1/20 the period of fastest oscillation in the simulated system. The C-H, N-H, and O-H vibrations have periods of order 10 -14 sec (= 10 fs). If the time unit is chosen as 1 tu = 10 -14, then the time step becomes on the order of 0.2 fs (0.02 tu). However, it is possible in Molecular Dynamics simulations to constrain the C-H, N-H, and O-H distances allowing the use of time steps up to 2 fs. The procedure involved is called SHAKE in which the H atoms are initially moved according to the algorithms given above. Then, since each bond is considered independently of another, all the C-H, N-H, and O-H distances are corrected such that the bond length is constrained within a given tolerance.
 = 1 x sec; 1 nanosecond (ns) = 1 x sec; 1000 ps = 1 ns). Molecular Dynamics is extremely useful to study properties that depend on high frequency vibrations. It is difficult to study properties such as rotations, diffusion mechanisms, and viscosity using Molecular Dynamics where local events are equilibrated on the order of 100 ps and longer. Unless small time steps are used, very significant errors in the energy will be introduced. In practice the time step is taken in femtoseconds (1 femtosecond (fs) = 1 x sec). To ensure numerical stability for the solution of the equations of motion, the time step must be at most 1/ /20 the period of fastest oscillation in the simulated system. The C-H, N-H, and O-H vibrations have periods of order sec (= 10 fs). If the time unit is chosen as 1 tu = , then the time step becomes on the order of 0.2 fs (0.02 tu). However, it is possible in Molecular Dynamics simulations to constrain the C-H, N-H, and O-H distances allowing the use of time steps up to 2 fs. The procedure involved is called SHAKE in which the H atoms are initially moved according to the algorithms given above. Then, since each bond is considered independently of another, all the C-H, N-H, and O-H distances are corrected such that the bond length is constrained within a given tolerance..")
15
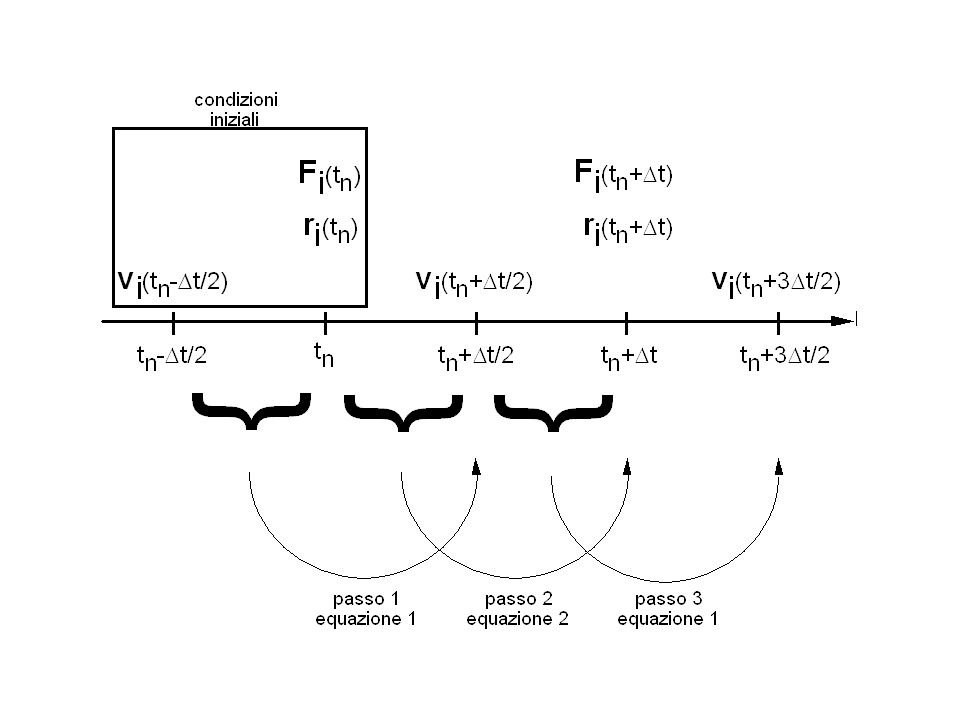
The `Leap Frog' algorithm sottraendo membro a membro: ovvero: equazione 1

16
Analogamente per la posizione: sottraendo membro a membro: ovvero: equazione 2

18
Verlet algorithm sommando membro a membro: dove: L’espressione di è corretta fino ai termini di ordine Per ricavare la velocità viene fatta l’approssimazione che rimanga costante durante :

20
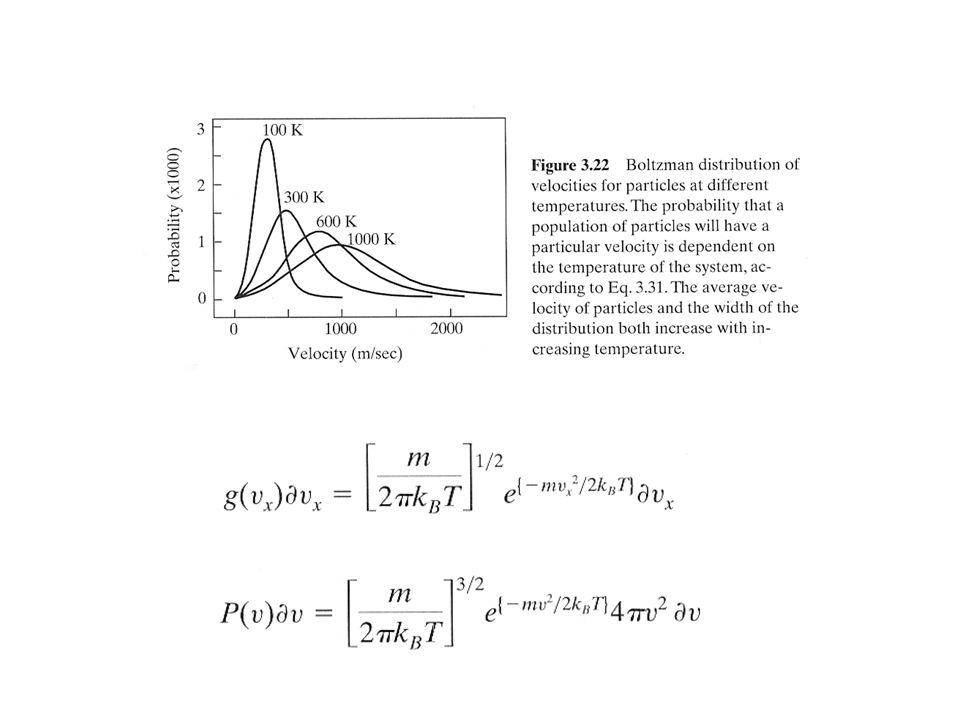
Performing a Simulation: Minimization, Heating, Equilibration, and Production Initially, the system should be minimized to eliminate the close atom-atom contacts, since this could produce large forces and consequently local strain. After the system is minimized, it is gradually heated to the desired temperature by assigning velocities to the atoms according to a Maxwell distribution. This is done periodically to the system to ensure that there are no local heating problems. After the system attains the desired temperature, it is important to go through an equilibration phase to allow for a redistribution of energy and produce stability. However, it may still be necessary to periodically rescale the velocities to bring the system to the desired temperature. The final phase involves production dynamics. In the Microcanical Ensemble (NVE dynamics) the system undergoes dynamics with no rescaling of the velocities. The temperature may start to drift as a result of a short cut-off for the long-range electrostatic forces.
 the system undergoes dynamics with no rescaling of the velocities. The temperature may start to drift as a result of a short cut-off for the long-range electrostatic forces..")
21
coordinate di input minimizzazione assegnazione delle velocità: rinormalizzazione delle velocità per avere momento lineare nullo calcolo forze calcolo nuove posizioni (=1 femtosecondo) normalizzazione delle velocità per mantenere T costante alcuni picosecondi calcolo nuove posizioni (=1 femtosecondo) alcuni picosecondi calcolo forze il sistema è stabile? sino { T stabile T uniforme Velocità maxwelliane T finale? sino continuazione simulazione memorizzazione di coordinate e velocità

23
Constrains V tot = V campo di forza + V constrains V constrains = A (grandezza calcolata-grandezza osservata) 2 grandezza NMR NOE X ray
 2 grandezza NMR NOE X ray")
25
Valutazione statistica dell’entropia Matrice di covarianza configurazionale

26
Funzione di correlazione: Autocorrelazione: Autocorrelazione della velocità:

27
Monte Carlo and Dynamic Simulations Methods Similarities Both the Monte Carlo (MC) and Stochastic Dynamics (SD) methods are thermodynamically equivalent ways to generate conformational states. Both methods are stochastic techniques which incorporate randomness in searching for microstates in phase space. SD assigns random initial velocities to every atom followed by deterministic classical mechanics. On the other hand, MC continually assigns random changes to the system by modifying a few degrees of freedom at a time. Both methods incorporate the concept of temperature and thermal fluctuations allowing higher energy structures and producing a wide spectrum of conformational states which may accurately reflect the experimental conditions. Differences In SD the next configuration is generated by moving the atoms in a deterministic manner as a result of evaluating the force defined by energy gradients. Each new structure is a unique result based on the forces acting on the previous structure. In the MC method, the atoms are moved randomly, and the next conformation is only a guess. The main difference between SD and MC is that SD uses forces and requires that the derivatives of the energy be calculated while MC requires only the calculation of the energy, E. Because SD integrates the classical equations of motion, it is quite suitable for studying time dependent phenomena. On the other hand, only E is needed for MC, and there is no concept of time.

Presentazioni simili















 Anche se Cassirer ritiene che la religione sia il naturale proseguimento del mito, dobbiamo trattare.>")


>")
>")
>")


