Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Giulio Pavesi Dipartimento di BioScienze Università di Milano
LID 2013 Bioinformatica Giulio Pavesi Dipartimento di BioScienze Università di Milano

2
Programma (di massima)
Storia evolutiva: ortologhi e paraloghi, identificazione di domini conservati (parziale ripasso dall’anno precedente) Struttura del gene: trascritti e splicing alternativi (parziale ripasso dall’anno precedente). Espressione del gene: housekeeping o tessuto specifica, misurata attraverso metodi tradizionali (microarray) e RNA-Seq, con identificazione di isoforme tessuto, sviluppo e condizione-specifiche. Preparazione di una validazione, se voglio validare la presenza di un particolare trascritto tramite PCR. Disegno di primer isoforma-specifici e loro test tramite e-PCR.
 Struttura del gene: trascritti e splicing alternativi (parziale ripasso dall’anno precedente). Espressione del gene: housekeeping o tessuto specifica, misurata attraverso metodi tradizionali (microarray) e RNA-Seq, con identificazione di isoforme tessuto, sviluppo e condizione-specifiche. Preparazione di una validazione, se voglio validare la presenza di un particolare trascritto tramite PCR. Disegno di primer isoforma-specifici e loro test tramite e-PCR.")
3
Programma (di massima)
Varianti: identificazione di SNPs e loro possible ruolo (in regione codificante:sinonime/non sinonime, introni/splicing alternativi, promotore) Regolazione della trascrizione del gene: struttura della cromatina, modificazioni istoniche, legame di fattori di trascrizione in promotori ed enhancer. Caratterizzazione di insiemi di geni, ovvero se prendo un gene e i suoi vicini di espressione (o altri sistemi di caratterizzazione, es. geni co-regolati con esso). Identificazione di categorie di gene ontology e pathways arricchiti.
 Regolazione della trascrizione del gene: struttura della cromatina, modificazioni istoniche, legame di fattori di trascrizione in promotori ed enhancer. Caratterizzazione di insiemi di geni, ovvero se prendo un gene e i suoi vicini di espressione (o altri sistemi di caratterizzazione, es. geni co-regolati con esso). Identificazione di categorie di gene ontology e pathways arricchiti.")
4
Strumenti

5
“Quaderno” In ogni lezione vi verrà presentato come rispondere alle domande mostrate in precedenza utilizzando strumenti bioinformatici Verranno utilizzati geni “di esempio” Dovrete ripetere per conto vostro le procedure mostrate sul gene che vi è stato assegnato come compito Dovrete descrivere sul “quaderno” le procedure utilizzate e i risultati ottenuti (anche, possibilmente, discutendoli) - utile fare “foto” (screen captures) dei diversi output ottenuti Esame: discutere il quaderno, le metodologie applicate e i relativi risultati con il prof. (io)
 - utile fare foto (screen captures) dei diversi output ottenuti. Esame: discutere il quaderno, le metodologie applicate e i relativi risultati con il prof. (io)")
6
Domanda 1 Struttura del gene: promotori, trascritti, e splicing alternativi (parziale ripasso dall’anno precedente). Espressione del gene: housekeeping o tessuto specifica, misurata attraverso metodi tradizionali (microarray) e RNA-Seq, con identificazione di isoforme tessuto, sviluppo e condizione-specifiche.
 e RNA-Seq, con identificazione di isoforme tessuto, sviluppo e condizione-specifiche.")
7
Il “gene” Le annotazioni “ufficiali” dei geni sono curate da appositi consorzi/enti/gruppi di ricerca, solitamente gli stessi che curano il sequenziamento del genoma. Esempi: D.melanogaster: “Flybase” A.thaliana: “TAIR” S.cerevisiae: “SGD“ Zebrafish: “ZGD” Uomo e altri mammiferi: diverse annotazioni

8
Annotare i geni Un gene viene annotato in una regione genomica che produce RNA - non necessariamente codificante L’annotazione viene compiuta “mappando” i trascritti sul genoma, e identificando le regioni da cui i trascritti stessi provengono I trascritti utilizzati vengono “catalogati” ufficialmente dal consorzio che effettua l’annotazione, con il gene corrispondente cui sono attribuiti Le annotazioni sono (per lo meno dichiarate essere) “curate manualmente”, ovvero vengono valutate, ad esempio: L’”affidabilità” dei trascritti utilizzati (completezza, assenza di errori di sequenziamento) L’”affidabilità” della mappatura/attribuzione al gene La presenza o meno di una regione codificante “affidabile” e la relativa proteina codificata
 curate manualmente , ovvero vengono valutate, ad esempio: L’ affidabilità dei trascritti utilizzati (completezza, assenza di errori di sequenziamento) L’ affidabilità della mappatura/attribuzione al gene. La presenza o meno di una regione codificante affidabile e la relativa proteina codificata.")
9
Annotare i geni Per alcuni genomi (es. uomo) sono disponibili diverse annotazioni che fondamentalmente differiscono per i trascritti utilizzati Annotazioni disponibili per l’uomo sul browser UCSC: Geni “RefSeq” (curati da NCBI) - pochi trascritti, ma buoni Geni “UCSC” - (curati da UCSC) più trascritti per gene, più splicing alternativi - meno “buoni” Geni “ENSEMBL” (curati da EBI) - più trascritti ancora - meno buoni - include trascritti parziali Geni “Gencode” (curati da ENCODE) - simili a ENSEMBL Alcune annotazioni includono solo geni “protein coding”, altre includono anche RNA non codificanti (es. microRNA, ma anche tRNA e rRNA)
 - pochi trascritti, ma buoni. Geni UCSC - (curati da UCSC) più trascritti per gene, più splicing alternativi - meno buoni Geni ENSEMBL (curati da EBI) - più trascritti ancora - meno buoni - include trascritti parziali. Geni Gencode (curati da ENCODE) - simili a ENSEMBL. Alcune annotazioni includono solo geni protein coding , altre includono anche RNA non codificanti (es. microRNA, ma anche tRNA e rRNA)")
10
Quale annotazione? ...tra le varie annotazioni disponibili (se più di una) quale usare? ...questione di gusti, e dipende da quello che si deve fare Annotazioni con pochi trascritti ma buoni (es. RefSeq) vanno benissimo per studiare le “caratteristiche generali” del gene (es. ipotesi sulla funzione della proteina codificata - posso usare quella annotata sul trascritto RefSeq) Annotazioni con più trascritti sono utili es. per studiare splicing alternativi e isoforme del gene
 vanno benissimo per studiare le caratteristiche generali del gene (es. ipotesi sulla funzione della proteina codificata - posso usare quella annotata sul trascritto RefSeq) Annotazioni con più trascritti sono utili es. per studiare splicing alternativi e isoforme del gene.")
11
Annotazione “next-generation”
La “bontà” o meno di un’annotazione dipende dalla disponibilità di trascritti Fino a pochi anni fa, le annotazioni venivano compiute a partire dalle sequenze disponibili (es. database “GenBank - Nucleotide” NCBI) prodotte da laboratori di tutto il mondo “Campionamento” del trascrittoma “biased”, ovvero per i geni più studiati (es. TP53, DMD in uomo) erano disponibili molti più trascritti di geni poco studiati o poco caratterizzati
 prodotte da laboratori di tutto il mondo. Campionamento del trascrittoma biased , ovvero per i geni più studiati (es. TP53, DMD in uomo) erano disponibili molti più trascritti di geni poco studiati o poco caratterizzati.")
12
Annotazione “next-generation”
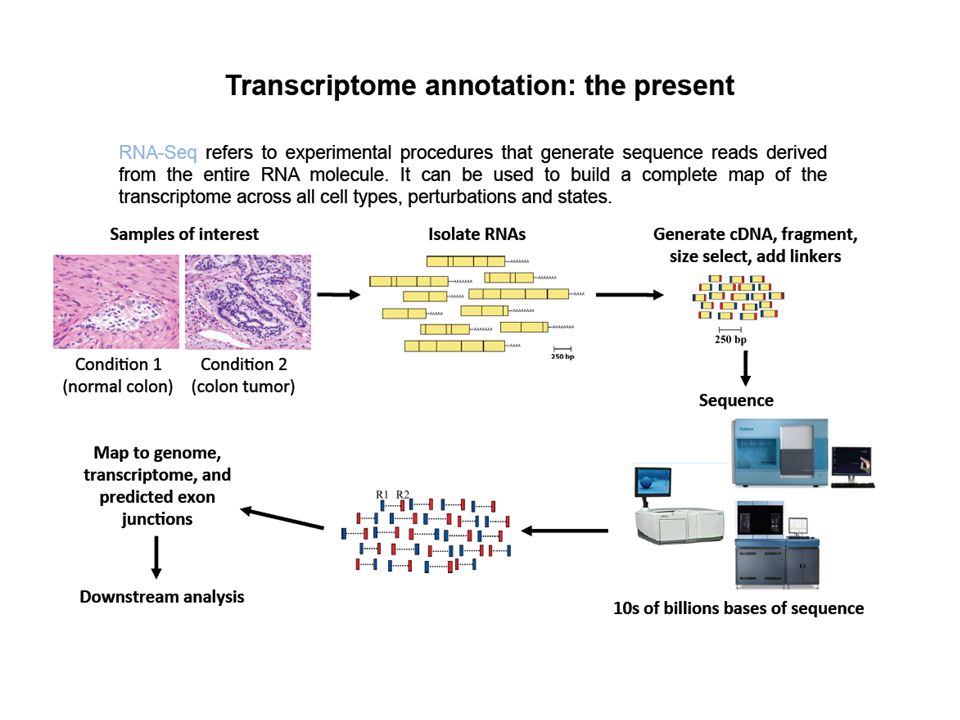
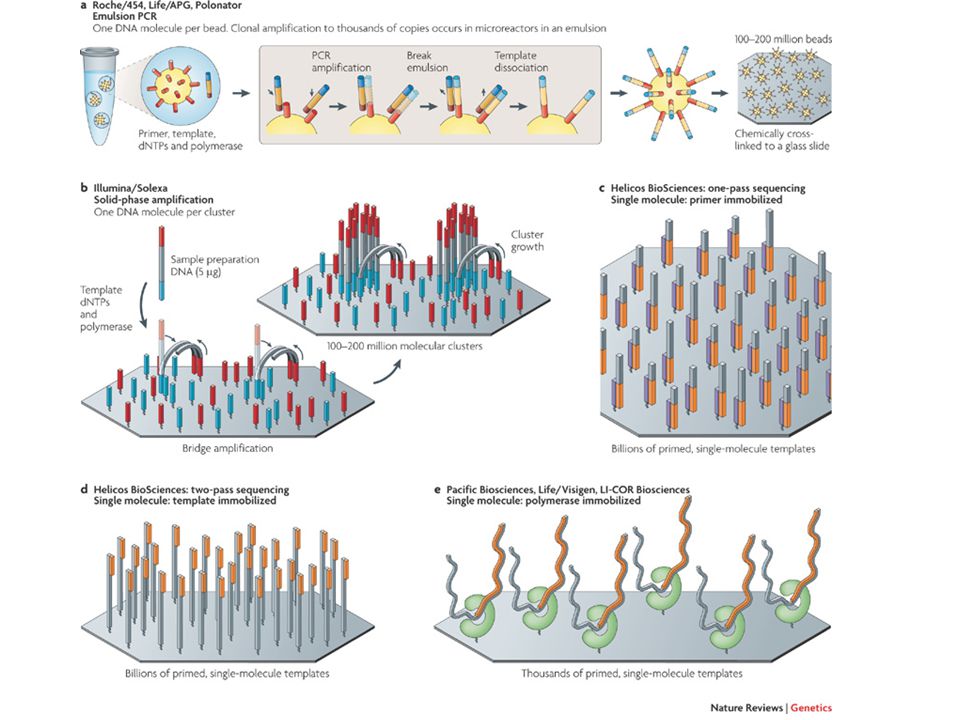
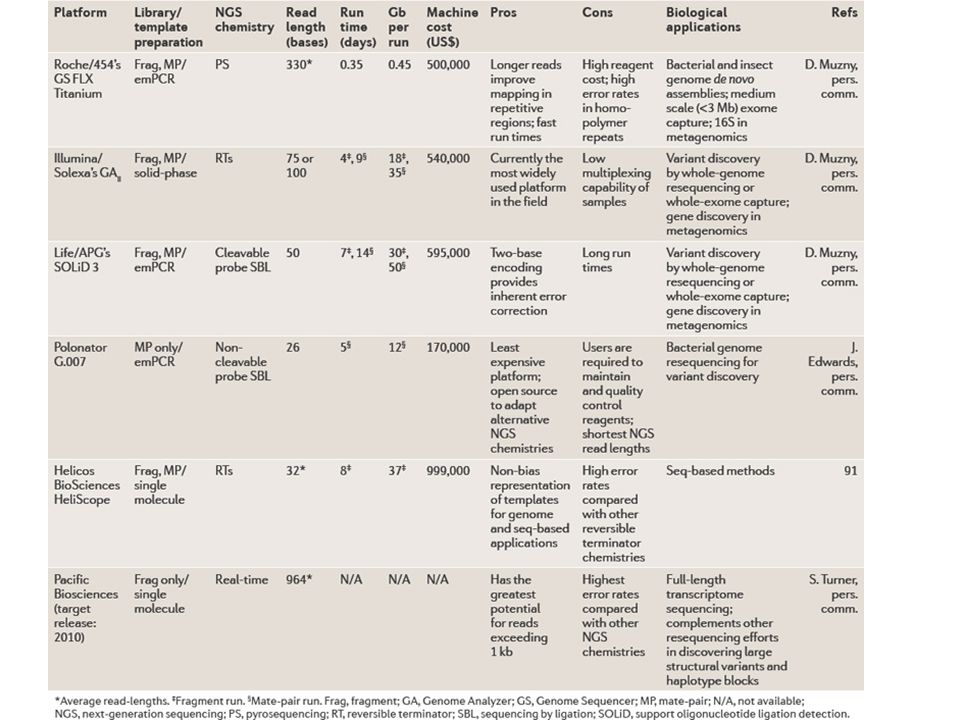
Con l’introduzione delle tecniche di sequenziamento dette “next-generation sequencing” la possibilità di sequenziare - a costi relativamente contenuti - interi genomi o trascrittomi è diventata accessibile anche a laboratori di dimensione/disponibilità economica piccolo/media Con “RNA-Seq” si intende l’applicazione di un metodo di sequenziamento “next-generation” al sequenziamento di un campione di RNA estratto da un particolare campione/linea cellulare/ecc. ecc.

16
RNA (cDNA) > 1000 bp Sanger sequencing (700 bp)
 > 1000 bp Sanger sequencing (700 bp)")
17
RNA (cDNA) > 1000 bp 50 bp 50 bp 50 bp 50 bp 50 bp 50 bp 50 bp
 > 1000 bp 50 bp 50 bp 50 bp 50 bp 50 bp 50 bp 50 bp")
19
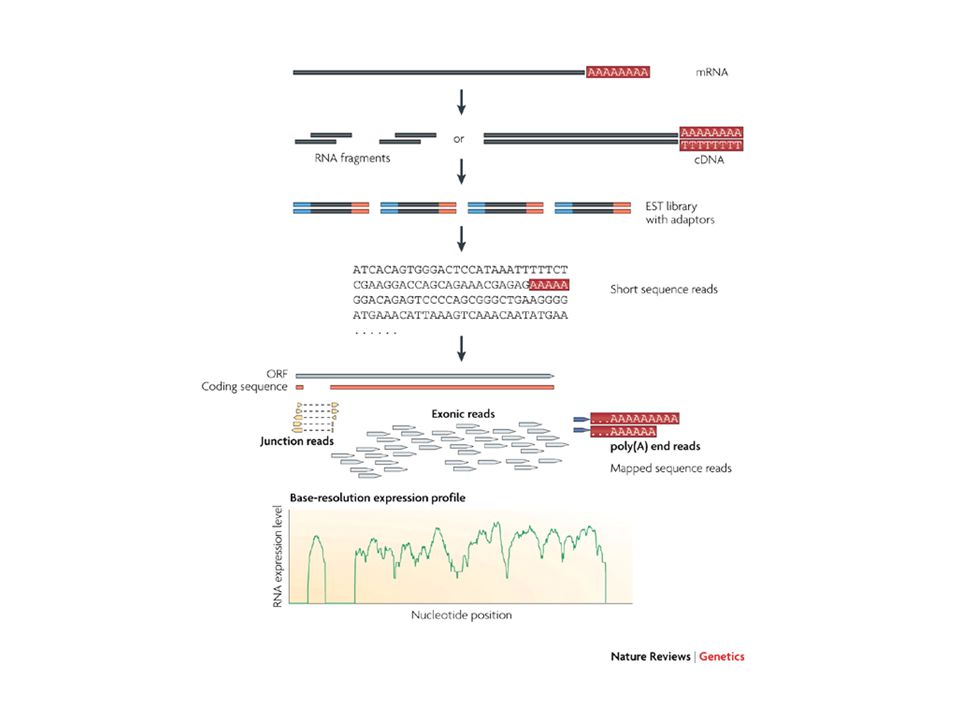
RNA-Seq per misurare l’”espressione”
Il campione originale di RNA è stato frammentato I frammenti (reads) effettivamente sequenziati vengono selezionati a caso dal sequenziatore La probabilità che un RNA abbia uno dei suoi frammenti selezionati e sequenziati è proporzionale all’abbondanza dell’RNA stesso nel campione iniziale Il “livello di trascritto” di un gene è quindi proporzionale al numero di frammenti (reads) assegnati ai suoi trascritti
 effettivamente sequenziati vengono selezionati a caso dal sequenziatore. La probabilità che un RNA abbia uno dei suoi frammenti selezionati e sequenziati è proporzionale all’abbondanza dell’RNA stesso nel campione iniziale. Il livello di trascritto di un gene è quindi proporzionale al numero di frammenti (reads) assegnati ai suoi trascritti.")
20
RNA-Seq per misurare l’”espressione”
Poiché i frammenti (read) possono essere assegnati ai singoli esoni di un gene, è possibile anche misurare il livello di trascritto a livello di singolo esone Ovvero, è possibile stimare il livello di “espressione” per ogni trascritto alternativo/isoforma del gene 1 2 3 10 10 10 Il livello di trascritto dei 3 esoni è uniforme ( ), quindi possiamo ipotizzare che il trascritto originale sia costituito dalla giunzione di 1-2-3
 possono essere assegnati ai singoli esoni di un gene, è possibile anche misurare il livello di trascritto a livello di singolo esone. Ovvero, è possibile stimare il livello di espressione per ogni trascritto alternativo/isoforma del gene Il livello di trascritto dei 3 esoni è uniforme ( ), quindi. possiamo ipotizzare che il trascritto originale sia costituito dalla. giunzione di")
21
RNA-Seq per misurare l’”espressione”
Poiché i frammenti (read) possono essere assegnati ai singoli esoni di un gene, è possibile anche misurare il livello di trascritto a livello di singolo esone Ovvero, è possibile stimare il livello di “espressione” per ogni trascritto alternativo/isoforma del gene 1 2 3 10 3 10 Il livello di trascritto dei 3 esoni NON è uniforme ( ). Gli esoni 1-3 (costitutivi) hanno livello 10 L’esone 2 (cassetta) ha livello 3 Ipotesi: l’esone 2 è incluso nel 30% dei trascritti maturi, e quindi:
 possono essere assegnati ai singoli esoni di un gene, è possibile anche misurare il livello di trascritto a livello di singolo esone. Ovvero, è possibile stimare il livello di espressione per ogni trascritto alternativo/isoforma del gene Il livello di trascritto dei 3 esoni NON è uniforme ( ). Gli esoni 1-3 (costitutivi) hanno livello 10 L’esone 2 (cassetta) ha livello 3. Ipotesi: l’esone 2 è incluso nel 30% dei trascritti maturi, e quindi:")
22
RNA-Seq per identificare splicing alternativi
1 2 3 Sappiamo che l’esone 2 è un potenziale esone cassetta. Viene incluso o meno nei trascritti maturi nella condizione che sto studiando? Potenzialmente sì (ci sono alcuni read che mappano sull’esone due - come avevamo visto nell’esempio precedente) Come ulteriore validazione è anche possibile: 1 2 3 10 3 10
 Come ulteriore validazione è anche possibile:")
23
RNA-Seq per identificare splicing alternativi
1 2 3 Se l’esone 2 è incluso, allora deve essere “unito” all’esone 1 (al 5’) e all’esone 3 (al 3’) Quindi, lo splicing predetto viene “confermato” se troviamo read che stanno “a cavallo” di due esoni. Quelli verdi confermano la “giunzione” tra esone 1-2 e esone 2-3; quello rosso mostra anche la giunzione alternativa 1-3 che corrisponde all’excisione dell’esone 2 1 2 3
 e all’esone 3 (al 3’) Quindi, lo splicing predetto viene confermato se troviamo read che stanno a cavallo di due esoni. Quelli verdi confermano la giunzione tra esone 1-2 e esone 2-3; quello rosso mostra anche la giunzione alternativa 1-3 che corrisponde all’excisione dell’esone")
24
RNA-Seq per misurare l’”espressione”
Poiché i frammenti (read) possono essere assegnati ai singoli esoni di un gene, è possibile anche misurare il livello di trascritto a livello di singolo esone Ovvero, è possibile stimare il livello di “espressione” per ogni trascritto alternativo/isoforma del gene Livello stimato: 3 1 3 Livello stimato: 7 1 2 3 Il livello di trascritto dei 3 esoni NON è uniforme ( ). Gli esoni 1-3 (costitutivi) hanno livello 10 L’esone 2 (cassetta) ha livello 3 Ipotesi: l’esone 2 è incluso nel 30% dei trascritti maturi.
 possono essere assegnati ai singoli esoni di un gene, è possibile anche misurare il livello di trascritto a livello di singolo esone. Ovvero, è possibile stimare il livello di espressione per ogni trascritto alternativo/isoforma del gene. Livello stimato: Livello stimato: Il livello di trascritto dei 3 esoni NON è uniforme ( ). Gli esoni 1-3 (costitutivi) hanno livello 10 L’esone 2 (cassetta) ha livello 3. Ipotesi: l’esone 2 è incluso nel 30% dei trascritti maturi.")
25
“Read Mapping” in RNA-Seq
5’ 3’ mRNA Reads 5’ 3’

26
RNA-Seq vs. Arrays Cassette exon 3’ 5’ A traditional “Chip”
Affy probe A traditional “Chip” might have probes designed on constitutive exons Impossible to tell whether the middle exon is spliced or not “Tiling” arrays and “exon arrays” have been introduced to overcome this problem

27
RNA-Seq Cassette exon spliced out 3’ 5’ 3’ 5’
Cassette exon not spliced out

28
RNA-Seq Cassette exon non incluso 5’ 3’ 5’ 3’ Cassette exon incluso

29
RNA-Seq: detecting KNOWN splicings
Build an “exon junction” library from existing transcript annotations (e.g. joining the sequence fragments in red), joining the 3’ with the 5’ of neighboring exons Each “exon junction” should be unique and of the same length of the sequenced reads Map reads that were “unmapped” (on the genome) on the junction library A B C D 5’ 3’ A-B C-D A-D
, joining the 3’ with the 5’ of neighboring exons. Each exon junction should be unique and of the same length of the sequenced reads. Map reads that were unmapped (on the genome) on the junction library. A. B. C. D. 5’ 3’ A-B. C-D. A-D.")
30
RNA-Seq: trovare NUOVI splicing
What if: We have reads mapping within an annotated intron? A D 5’ 3’ ?? A-D

31
RNA-Seq: detecting NOVEL splicings
Define possible exon boundaries looking for canonical splice sites and see whether you can join it to annotated exons by splitting unmapped sequence reads 5’ 3’ GT AG GT AG

32
RNA-Seq: using PAIRED-END sequences
200 bp 5’ 3’ 5’ 3’

33
RNA-Seq: using PAIRED-END sequences
Easier to detect novel exons and join them to existing annotations 5’ 3’ ???? Novel exon

34
RNA-Seq: using PAIRED-END sequences
Easier (computationally) to assign to their location the paired unmapped reads 5’ 3’ ????
 to assign to their location. the paired unmapped reads. 5’ 3’")
Presentazioni simili


>")













 Piccole molecole di RNA (20-22 nt)>")



