Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Facoltàdi Medicina e Chirurgia POLO C Diletta Mauro Daniele Messina Luca Petrella Isabella Pollicina Prof. C. Di Pietro

2
Malattie legate a mutazioni strutturali dei cromosomi

3
Mutazioni Una mutazione è definibile come un evento casuale e stabile che produce un cambiamento del patrimonio genetico ed è quindi ereditabile.

4
Mutazioni Mutazioni Genomiche Geniche Cromosomiche Cromosomiche

5
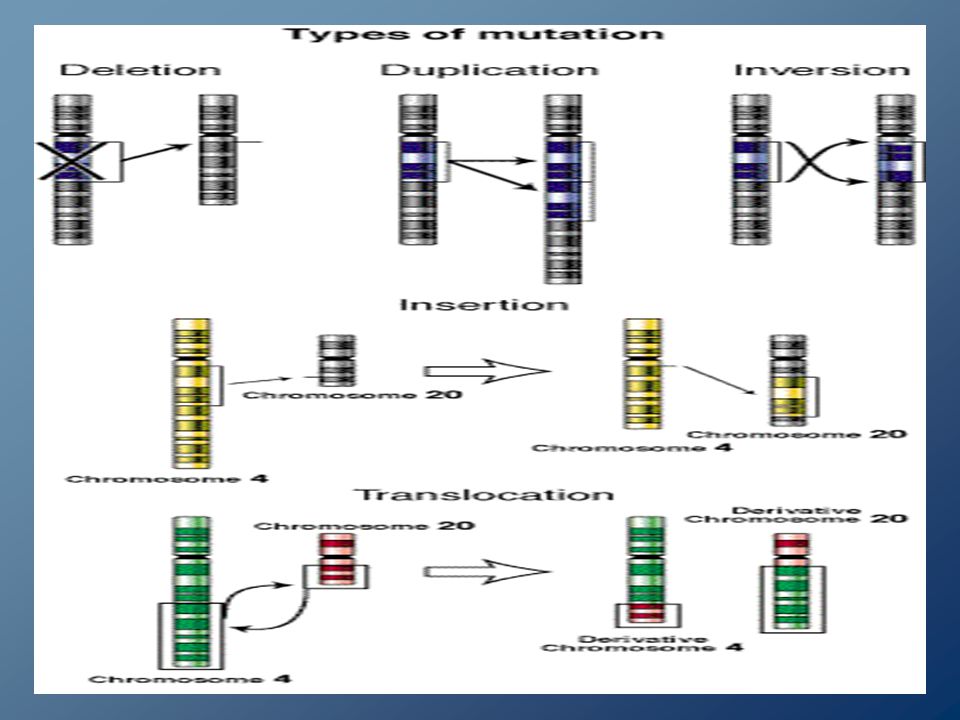
Mutazioni strutturali dei Cromosomi DUPLICAZIONE: In un cromosoma sono presenti più copie di uno stesso segmento cromosomico. DELEZIONE: Si ha la perdita di frammenti di un cromosoma. Possono essere terminali o interstiziali. INVERSIONE: All’interno di uno stesso cromosoma si formano due fratture e il frammento exciso è inserito dopo aver effettuato una rotazione di circa 180°. TRASLOCAZIONE: Si ha lo spostamento di tratti di dna, secondo almeno tre possibilità Traslocazione intracromosomica non reciproca; Traslocazione intercromosomica non reciproca; Traslocazione intercromosomica reciproca.

6
Esempi di inversione Pericentrica (a) e Paracentrica (b)
 e Paracentrica (b)")
8
Durante il crossing-over, i cromosomi che hanno subito inversione assumono questa caratteristica configurazione a “LOOP”.

9
Si possono inoltre avere: Si possono inoltre avere: CROMOSOMI AD ANELLO CROMOSOMI AD ANELLO (Ring Chromosomes); (Ring Chromosomes); ISOCROMOSOMI; ISOCROMOSOMI; TRASLOCAZIONI ROBERTSONIANE TRASLOCAZIONI ROBERTSONIANE Traslocazioni non reciproche dovute al trasferimento di un cromosoma sul centromero di un cromosoma Traslocazioni non reciproche dovute al trasferimento di un cromosoma sul centromero di un cromosoma acrocentrico. acrocentrico.

11
Malattie legate a DELEZIONI Esempi di patologie umane legate a delezioni cromosomiche sono: Esempi di patologie umane legate a delezioni cromosomiche sono: 1) Sindrome del cri-du-chat; 2) Sindrome di Prader-Willi-Angelman; 3) Sindrome di Miller-Dieker
 Sindrome del cri-du-chat; 2) Sindrome di Prader-Willi-Angelman; 3) Sindrome di Miller-Dieker")
12
1) Sindrome del cri-du-chat Cri du chat è il nome che il genetista francese Jerome Léjeune ha dato nel 1963 ad una sindrome riconoscibile fin dalla nascita a causa del vagito acuto e flebile come un miagolio. Si tratta di una malattia rara; l'incidenza è compresa fra 1/15.000 e 1/50.000 nati vivi, anche se è una delle più comuni sindromi da delezione nell'uomo.

13
I neonati hanno alcune particolarità dei lineamenti che li rendono somiglianti fra loro: faccia tonda occhi che sembrano distanti a causa dell’epicanto mandibola piccola e mento sfuggente (microretrognazia) basso peso, ipotonia difficoltà di suzione Crescendo, i tratti cambiano: il volto si allunga, è frequente lo strabismo divergente, mani e piedi sono piccoli. La statura e il peso sono generalmente inferiori alla norma. La voce conserva un caratteristico timbro acuto. Si verifica MICROCEFALIA.

14
E’ causata dalla delezione di un frammento del braccio corto di uno dei cromosomi 5 (5p-). La delezione può coinvolgere la parte terminale o una parte all’interno (interstiziale) del braccio corto. E’ causata dalla delezione di un frammento del braccio corto di uno dei cromosomi 5 (5p-). La delezione può coinvolgere la parte terminale o una parte all’interno (interstiziale) del braccio corto. Gli studi cromosomici suggeriscono la presenza di "regioni critiche" che, quando incluse nella delezione, sono responsabili delle manifestazioni tipiche della sindrome. In queste regioni critiche sono stati individuati due geni, Semaforina e δ- catenina, che potrebbero essere coinvolti nello sviluppo cerebrale. Gli studi cromosomici suggeriscono la presenza di "regioni critiche" che, quando incluse nella delezione, sono responsabili delle manifestazioni tipiche della sindrome. In queste regioni critiche sono stati individuati due geni, Semaforina e δ- catenina, che potrebbero essere coinvolti nello sviluppo cerebrale.
 del braccio corto. E’ causata dalla delezione di un frammento del braccio corto di uno dei cromosomi 5 (5p-). La delezione può coinvolgere la parte terminale o una parte all’interno (interstiziale) del braccio corto. Gli studi cromosomici suggeriscono la presenza di regioni critiche che, quando incluse nella delezione, sono responsabili delle manifestazioni tipiche della sindrome. In queste regioni critiche sono stati individuati due geni, Semaforina e δ- catenina, che potrebbero essere coinvolti nello sviluppo cerebrale. Gli studi cromosomici suggeriscono la presenza di regioni critiche che, quando incluse nella delezione, sono responsabili delle manifestazioni tipiche della sindrome. In queste regioni critiche sono stati individuati due geni, Semaforina e δ- catenina, che potrebbero essere coinvolti nello sviluppo cerebrale..")
15
Bambini affetti dalla sindrome del Cri- du-Chat che mostrano le particolarità nei lineamenti tipiche della malattia. Cariotipo di un individuo affetto da cri-du-chat.

16
Problemi medici Problemi medici Nel periodo neonatale: problemi respiratori e difficoltà di suzione; Nel periodo neonatale: problemi respiratori e difficoltà di suzione; Malformazioni: cardiache e renali (non frequenti), ernie inguinali, sindattilia, lussazione congenita delle anche, piede piatto o equino-varo, ipospadia, criptorchidismo; Malformazioni: cardiache e renali (non frequenti), ernie inguinali, sindattilia, lussazione congenita delle anche, piede piatto o equino-varo, ipospadia, criptorchidismo; Vari problemi oculistici e ortopedici; Vari problemi oculistici e ortopedici; Anomalie della laringe. Anomalie della laringe. Sopravvivenza Sopravvivenza Prolungata, diversi soggetti hanno superato i 60 anni di età. Mortalità di circa il 10%. Prolungata, diversi soggetti hanno superato i 60 anni di età. Mortalità di circa il 10%.

17
Trattamenti riabilitativi-Terapie Non esiste una cura in senso stretto (farmacologica o chirurgica) per la sindrome del Cri du Chat. Il segmento di DNA perduto non è recuperabile e in ogni caso il danno cerebrale si verifica molto precocemente. E’ tuttavia possibile agire sulle conseguenze che l'alterazione genetica comporta mediante interventi riabilitativi che è importante intraprendere precocemente, fin dalle prime settimane di vita.

18
2) Sindrome di Prader-Willi-Angelman La sindrome di Prader Willi è una malattia genetica rara caratterizzata dall'alterazione del cromosoma 15. La sindrome di Prader Willi è una malattia genetica rara caratterizzata dall'alterazione del cromosoma 15. I soggeti affetti sono privi del senso di sazietà; la malattia inoltre è causa di una disfunzione nel metabolismo, che riduce la capacità dell'organismo di bruciare calorie assunte con I soggeti affetti sono privi del senso di sazietà; la malattia inoltre è causa di una disfunzione nel metabolismo, che riduce la capacità dell'organismo di bruciare calorie assunte con l'alimentazione. l'alimentazione.

19
Eziologia Eziologia La Prader-Willi è la più comune tra le sindromi di microdelezione cromosomica. Avviene per due diverse cause accertate, di tipo genetico: La Prader-Willi è la più comune tra le sindromi di microdelezione cromosomica. Avviene per due diverse cause accertate, di tipo genetico: Delezione paterna di una regione (contenente un gene o un gruppo di geni) normalmente sottoposta a imprinting, e in particolare attiva nel padre e imprintata nella madre; Delezione paterna di una regione (contenente un gene o un gruppo di geni) normalmente sottoposta a imprinting, e in particolare attiva nel padre e imprintata nella madre; Disomia uniparentale materna (ossia presenza di due copie di origine materna dello stesso cromosoma, le quali possono essere uguali o diverse). Disomia uniparentale materna (ossia presenza di due copie di origine materna dello stesso cromosoma, le quali possono essere uguali o diverse).
 normalmente sottoposta a imprinting, e in particolare attiva nel padre e imprintata nella madre; Delezione paterna di una regione (contenente un gene o un gruppo di geni) normalmente sottoposta a imprinting, e in particolare attiva nel padre e imprintata nella madre; Disomia uniparentale materna (ossia presenza di due copie di origine materna dello stesso cromosoma, le quali possono essere uguali o diverse). Disomia uniparentale materna (ossia presenza di due copie di origine materna dello stesso cromosoma, le quali possono essere uguali o diverse)..")
20
Nella PWS il gene materno è silenziato perché sotto imprinting, mentre quello paterno è deleto. Nella PWS il gene materno è silenziato perché sotto imprinting, mentre quello paterno è deleto. Il gene in questione è del cromosoma 15, nella regione 15q11-q13. Il gene in questione è del cromosoma 15, nella regione 15q11-q13. In questa patologia viene quindi a mancare il contributo paterno, e si avranno una serie di disturbi derivati dalla mancanza delle proteine da esso derivanti. In questa patologia viene quindi a mancare il contributo paterno, e si avranno una serie di disturbi derivati dalla mancanza delle proteine da esso derivanti. La regione deleta contiene le informazioni codificanti per la proteina umana necdin o necdina (NDNlocus), vicina alla regione centromerica della delezione, tra i due geni ZNF127 e SNRPN, entrambi sotto imprinting. La regione deleta contiene le informazioni codificanti per la proteina umana necdin o necdina (NDNlocus), vicina alla regione centromerica della delezione, tra i due geni ZNF127 e SNRPN, entrambi sotto imprinting.
, vicina alla regione centromerica della delezione, tra i due geni ZNF127 e SNRPN, entrambi sotto imprinting. La regione deleta contiene le informazioni codificanti per la proteina umana necdin o necdina (NDNlocus), vicina alla regione centromerica della delezione, tra i due geni ZNF127 e SNRPN, entrambi sotto imprinting..")
21
Sintomatologia Sintomatologia Ipotonia marcata che va via via scomparendo con l'età adolescenziale; Ipotonia marcata che va via via scomparendo con l'età adolescenziale; Appetito insaziabile; Appetito insaziabile; Possibile ritardo mentale; Possibile ritardo mentale; Ipogonadismo; Ipogonadismo; Strabismo; Strabismo; Mani e piedi piccoli; Mani e piedi piccoli; Iperfagia. Iperfagia.

22
Terapie Terapie I bambini PW vengono aiutati con la somministrazione dell‘ORMONE GH della crescita che fa in modo di dare più vitalità, di farli crescere in maniera corretta e di limitare le loro disfunzioni metaboliche che li spingono ad aumentare di peso più facilmente rispetto ai soggetti sani. I bambini PW vengono aiutati con la somministrazione dell‘ORMONE GH della crescita che fa in modo di dare più vitalità, di farli crescere in maniera corretta e di limitare le loro disfunzioni metaboliche che li spingono ad aumentare di peso più facilmente rispetto ai soggetti sani.

23
3) Sindrome di Miller-Dieker 3) Sindrome di Miller-Dieker Incidenza: 7 nati su milione, anche se probabilmente sottostimata. Incidenza: 7 nati su milione, anche se probabilmente sottostimata. Patogenesi: Microdelezione de novo nella regione 17p13.3 nell’8% dei casi, mentre il 20 % ha ereditato la delezione da uno dei genitori portatore di un riarrangiamento cromosomico bilanciato. Il gene LIS1, presente nel tratto deleto, è stato associato alle tipiche caratteristiche fenotipiche della sindrome. Patogenesi: Microdelezione de novo nella regione 17p13.3 nell’8% dei casi, mentre il 20 % ha ereditato la delezione da uno dei genitori portatore di un riarrangiamento cromosomico bilanciato. Il gene LIS1, presente nel tratto deleto, è stato associato alle tipiche caratteristiche fenotipiche della sindrome.

24
Fenotipo e storia naturale: Disgenesia cerebrale, epilessia,disturbi all’alimentazione, ipotonia, lento accrescimento e dimorfismi facciali. Fenotipo e storia naturale: Disgenesia cerebrale, epilessia,disturbi all’alimentazione, ipotonia, lento accrescimento e dimorfismi facciali. Trattamento: Per evitare le complicazioni legate ai problemi alimentari e della deglutizione si possono usare tubi nasogastrici e si può effettuare gastrostomia (soluzione a lungo termine). Infatti la Miller-Dieker è incurabile e la terapia è focalizzata su trattamenti palliativi. Trattamento: Per evitare le complicazioni legate ai problemi alimentari e della deglutizione si possono usare tubi nasogastrici e si può effettuare gastrostomia (soluzione a lungo termine). Infatti la Miller-Dieker è incurabile e la terapia è focalizzata su trattamenti palliativi.
. Infatti la Miller-Dieker è incurabile e la terapia è focalizzata su trattamenti palliativi. Trattamento: Per evitare le complicazioni legate ai problemi alimentari e della deglutizione si possono usare tubi nasogastrici e si può effettuare gastrostomia (soluzione a lungo termine). Infatti la Miller-Dieker è incurabile e la terapia è focalizzata su trattamenti palliativi..")
25
Bambini di età differenti affetti dalla sindrome di Miller-Dieker

26
Altre importanti delezioni sono state osservate in specifici tumori come: Altre importanti delezioni sono state osservate in specifici tumori come: 1) Retinoblastoma (delezione 13q14); 2) Tumore di Wilms (delezione 11q).
 Retinoblastoma (delezione 13q14); 2) Tumore di Wilms (delezione 11q).")
27
1) Retinoblastoma Il retinoblastoma è una patologia oculare e rappresenta il tumore maligno con maggiore diffusione in età pediatrica. Il retinoblastoma è una patologia oculare e rappresenta il tumore maligno con maggiore diffusione in età pediatrica. Essa deriva da una mutazione genetica che comporta la perdita di funzione di una proteina che funziona da oncosoppressore, la proteina Rb. Essa deriva da una mutazione genetica che comporta la perdita di funzione di una proteina che funziona da oncosoppressore, la proteina Rb. Esistono due tipi di patologia: Esistono due tipi di patologia: retinoblastoma ereditario retinoblastoma ereditario retinoblastoma sporadico. retinoblastoma sporadico. E’ importante sottolineare come la malattia si sviluppi soltanto quando entrambe le copie del gene perdono la loro funzione. E’ importante sottolineare come la malattia si sviluppi soltanto quando entrambe le copie del gene perdono la loro funzione.

28
RETINOBLASTOMA EREDITARIO (40%) RETINOBLASTOMA EREDITARIO (40%) dovuto all’eredità di un allele mutato, trasmesso alla progenie con le modalità tipiche dell’ereditarietà autosomica dominante di tipo mendeliano dovuto all’eredità di un allele mutato, trasmesso alla progenie con le modalità tipiche dell’ereditarietà autosomica dominante di tipo mendeliano RETINOBLASTOMA SPORADICO (60%) RETINOBLASTOMA SPORADICO (60%) Non è influenzato da componenti genetiche; ha decorso più lento Non è influenzato da componenti genetiche; ha decorso più lento FORMA BILATERALE FORMA UNILATERALE
 RETINOBLASTOMA EREDITARIO (40%) dovuto all’eredità di un allele mutato, trasmesso alla progenie con le modalità tipiche dell’ereditarietà autosomica dominante di tipo mendeliano dovuto all’eredità di un allele mutato, trasmesso alla progenie con le modalità tipiche dell’ereditarietà autosomica dominante di tipo mendeliano RETINOBLASTOMA SPORADICO (60%) RETINOBLASTOMA SPORADICO (60%) Non è influenzato da componenti genetiche; ha decorso più lento Non è influenzato da componenti genetiche; ha decorso più lento FORMA BILATERALE FORMA UNILATERALE")
29
Se il retinoblastoma non viene curato,la dimensione dell'occhio aumenta e il tumore inferisce su altre parti del corpo. Se il retinoblastoma non viene curato,la dimensione dell'occhio aumenta e il tumore inferisce su altre parti del corpo. Le metastasi precoci al fegato o ai polmoni sono frequenti. Le metastasi precoci al fegato o ai polmoni sono frequenti.

30
Proteina Rb Proteina Rb La proteina del retinoblastoma La proteina del retinoblastoma (pRb o Rb) è una proteina soppressore tumorale che è stata trovata non funzionante in numerosi tipi di cancro. Rb fu così chiamata perché fu scoperto che il retinoblastoma si sviluppava quando entrambi gli alleli del gene RB1 (il gene che codifica la proteina Rb) erano inattivati da una mutazione. La normale funzione di Rb è di agire come freno del ciclo cellulare, inibendo la crescita cellulare. (pRb o Rb) è una proteina soppressore tumorale che è stata trovata non funzionante in numerosi tipi di cancro. Rb fu così chiamata perché fu scoperto che il retinoblastoma si sviluppava quando entrambi gli alleli del gene RB1 (il gene che codifica la proteina Rb) erano inattivati da una mutazione. La normale funzione di Rb è di agire come freno del ciclo cellulare, inibendo la crescita cellulare.
 erano inattivati da una mutazione. La normale funzione di Rb è di agire come freno del ciclo cellulare, inibendo la crescita cellulare. (pRb o Rb) è una proteina soppressore tumorale che è stata trovata non funzionante in numerosi tipi di cancro. Rb fu così chiamata perché fu scoperto che il retinoblastoma si sviluppava quando entrambi gli alleli del gene RB1 (il gene che codifica la proteina Rb) erano inattivati da una mutazione. La normale funzione di Rb è di agire come freno del ciclo cellulare, inibendo la crescita cellulare..")
31
In molti casi, nel retinoblastoma è stata osservata una delezione a carico del braccio lungo del cromosoma 13, delezione che interessa il gene RB1, che codifica per pRb. In molti casi, nel retinoblastoma è stata osservata una delezione a carico del braccio lungo del cromosoma 13, delezione che interessa il gene RB1, che codifica per pRb. La delezione del gene RB1 in entrambe le copie alleliche causa l’assenza di pRb: La delezione del gene RB1 in entrambe le copie alleliche causa l’assenza di pRb: 1) impossibilità di errestare il ciclo cellulare; 1) impossibilità di errestare il ciclo cellulare; 2)origine di una neoplasia maligna. 2)origine di una neoplasia maligna.
 impossibilità di errestare il ciclo cellulare; 1) impossibilità di errestare il ciclo cellulare; 2)origine di una neoplasia maligna. 2)origine di una neoplasia maligna..")
32
Sintomatologia Si menifesta entro i primi 5 anni di vita Si menifesta entro i primi 5 anni di vita Segni principali: LEUCOCORIA, GLAUCOMA. Segni principali: LEUCOCORIA, GLAUCOMA. Effetto del flash fotografico su un occhio affetto da retinoblastoma

33
Terapie disponibili Terapie disponibili Esistono dei protocolli terapeutici che impongono determinati trattamenti a seconda dello stadio di malattia. I trattamenti locali includono: Esistono dei protocolli terapeutici che impongono determinati trattamenti a seconda dello stadio di malattia. I trattamenti locali includono: laser laser fotocoagulazione fotocoagulazione crioterapia crioterapia termoterapia transpupillare termoterapia transpupillare brachiterapia. brachiterapia. A questi trattamenti viene associata una chemioterapia per via sistemica o per via arteriosa. Nelle forme più avanzate di malattia è necessario rimuovere chirurgicamente il bulbo oculare malato. A questi trattamenti viene associata una chemioterapia per via sistemica o per via arteriosa. Nelle forme più avanzate di malattia è necessario rimuovere chirurgicamente il bulbo oculare malato.

34
2) Tumore di Wilms Il tumore di Wilms è un tumore pediatrico che può manifestarsi sporadicamente o in bambini con sindromi congenite. Circa il 10% dei bambini con tumore di Wilms presenta neoplasia bilaterale, fatto evidenziante l'esistenza di una lesione genetica predisponente. Il tumore di Wilms è un tumore pediatrico che può manifestarsi sporadicamente o in bambini con sindromi congenite. Circa il 10% dei bambini con tumore di Wilms presenta neoplasia bilaterale, fatto evidenziante l'esistenza di una lesione genetica predisponente.

35
Segni e sintomi manifestazione più frequente: massa addominale o al fianco, spesso asintomatica; manifestazione più frequente: massa addominale o al fianco, spesso asintomatica; dolore addominale; dolore addominale; vomito; vomito; ipertensione nel 60% dei pazienti. ipertensione nel 60% dei pazienti. Aspetto istologico Aspetto istologico La configurazione al microscopio del sottotipo ad istologia favorevole del tumore di Wilms è trifasica con elementi epiteliali, blastemali e stromali, simili a glomeruli abortivi. L'anaplasia viene riscontrata solo in circa il 10% dei casi, rappresentanti però il 60% dei decessi. Questo sottotipo istologico sfavorevole, che predilige pazienti in età più avanzata e non di razza bianca, presenta cellule di dimensioni 3 volte quelle normali, con nuclei ipercromici e mitosi anomale. La configurazione al microscopio del sottotipo ad istologia favorevole del tumore di Wilms è trifasica con elementi epiteliali, blastemali e stromali, simili a glomeruli abortivi. L'anaplasia viene riscontrata solo in circa il 10% dei casi, rappresentanti però il 60% dei decessi. Questo sottotipo istologico sfavorevole, che predilige pazienti in età più avanzata e non di razza bianca, presenta cellule di dimensioni 3 volte quelle normali, con nuclei ipercromici e mitosi anomale.

36
Nefroblastoma (tumore di Wilms)
")
37
Stadiazione Stadiazione I tumori di stadio I sono quelli limitati al rene e possono essere asportati completamente mantenendo intatta la superficie capsulare. Il tumore di stadio II si estende oltre il rene, ma può essere completamente escisso. Nel tumore di stadio III si ha l'estensione residua non ematogena della neoplasia, dopo l'intervento chirurgico, è limitata all'addome. Lo stadio IV indica la presenza di metastasi per via ematica, più frequentemente a carico del polmone. Nel 5-10% dei casi, si osserva un coinvolgimento renale bilaterale, di solito sincrono ( stadio V ). I tumori di stadio I sono quelli limitati al rene e possono essere asportati completamente mantenendo intatta la superficie capsulare. Il tumore di stadio II si estende oltre il rene, ma può essere completamente escisso. Nel tumore di stadio III si ha l'estensione residua non ematogena della neoplasia, dopo l'intervento chirurgico, è limitata all'addome. Lo stadio IV indica la presenza di metastasi per via ematica, più frequentemente a carico del polmone. Nel 5-10% dei casi, si osserva un coinvolgimento renale bilaterale, di solito sincrono ( stadio V ).
. I tumori di stadio I sono quelli limitati al rene e possono essere asportati completamente mantenendo intatta la superficie capsulare. Il tumore di stadio II si estende oltre il rene, ma può essere completamente escisso. Nel tumore di stadio III si ha l estensione residua non ematogena della neoplasia, dopo l intervento chirurgico, è limitata all addome. Lo stadio IV indica la presenza di metastasi per via ematica, più frequentemente a carico del polmone. Nel 5-10% dei casi, si osserva un coinvolgimento renale bilaterale, di solito sincrono ( stadio V )..")
38
Eziologia Delezioni geniche coinvolgenti almeno uno di due loci sul cromosoma 11 sono state riscontrate nelle cellule nel 33% dei tumori di Wilms. Le delezioni emizigotiche costituzionali di uno di questi loci, 11p13, sono anche associate a due rare sindromi: 1) sindrome WAGR 1) sindrome WAGR 2) sindrome Denys-Drash. 2) sindrome Denys-Drash. Inoltre, più dell'85% dei pazienti con tumore di Wilms con anaplasia hanno una mutazione nel gene soppressore P53. Inoltre, più dell'85% dei pazienti con tumore di Wilms con anaplasia hanno una mutazione nel gene soppressore P53.
 sindrome WAGR 1) sindrome WAGR 2) sindrome Denys-Drash. 2) sindrome Denys-Drash. Inoltre, più dell 85% dei pazienti con tumore di Wilms con anaplasia hanno una mutazione nel gene soppressore P53. Inoltre, più dell 85% dei pazienti con tumore di Wilms con anaplasia hanno una mutazione nel gene soppressore P53..")
39
Diagnosi La diagnosi di tumore di Wilms deve essere sospettata in qualsiasi bambino con massa addominale. La TAC offre molti vantaggi nella valutazione di un eventuale tumore di Wilms. Con la TAC il tumore di Wilms, di solito, viene visualizzato come una massa disomogenea di origine renale, con aree di minor densità indicanti necrosi. Diagnosi La diagnosi di tumore di Wilms deve essere sospettata in qualsiasi bambino con massa addominale. La TAC offre molti vantaggi nella valutazione di un eventuale tumore di Wilms. Con la TAC il tumore di Wilms, di solito, viene visualizzato come una massa disomogenea di origine renale, con aree di minor densità indicanti necrosi.

40
Terapia Il trattamento di prima linea dei tumori unilaterali è rappresentato dall'asportazione chirurgica del rene affetto attraverso un'incisione al fianco, anche in caso di presenza di metastasi polmonari. Si deve tentare in tutti i modi di asportare il tumore evitando la sua disseminazione; tuttavia, poiché la chemioterapia e la radioterapia postoperatorie sono in grado di distruggere i residui tumorali, non si dovrebbe procedere alla rimozione completa della neoplasia, se questo comporta rischi notevoli per il paziente. Terapia Il trattamento di prima linea dei tumori unilaterali è rappresentato dall'asportazione chirurgica del rene affetto attraverso un'incisione al fianco, anche in caso di presenza di metastasi polmonari. Si deve tentare in tutti i modi di asportare il tumore evitando la sua disseminazione; tuttavia, poiché la chemioterapia e la radioterapia postoperatorie sono in grado di distruggere i residui tumorali, non si dovrebbe procedere alla rimozione completa della neoplasia, se questo comporta rischi notevoli per il paziente.

41
Malattie legate a Traslocazioni Esempi di patologie legate a traslocazione cromosomica sono: Esempi di patologie legate a traslocazione cromosomica sono: 1) Linfoma di Burkitt; 2) Leucemia mieloide cronica; 3) Sindrome di Down familiare.
 Linfoma di Burkitt; 2) Leucemia mieloide cronica; 3) Sindrome di Down familiare.")
42
1) Linfoma di Burkitt Storia Storia Prende il nome da Denis Burkitt, un chirurgo che lavorava in Uganda e per primo lo descrisse nel 1958. Nel 1962 venne incluso nel gruppo dei linfomi maligni e negli anni successivi fu possibile appurare che una parte dei linfomi pediatrici negli Stati Uniti e in Europa erano istologicamente indistinguibili dal linfoma di Burkitt descritto in Africa. Si rese quindi necessario classificare nuovo gruppo di linfomi, che furono chiamati linfomi a piccole cellule non-clivate. Prende il nome da Denis Burkitt, un chirurgo che lavorava in Uganda e per primo lo descrisse nel 1958. Nel 1962 venne incluso nel gruppo dei linfomi maligni e negli anni successivi fu possibile appurare che una parte dei linfomi pediatrici negli Stati Uniti e in Europa erano istologicamente indistinguibili dal linfoma di Burkitt descritto in Africa. Si rese quindi necessario classificare nuovo gruppo di linfomi, che furono chiamati linfomi a piccole cellule non-clivate.

43
Caratteristiche Caratteristiche Il linfoma di Burkitt è una traslocazione intercromosomica reciproca che coinvolge i cromosomi 8 e 14, determinando l’attivazione dell’oncogene c- Myc sito sul cromosoma 8. Il linfoma di Burkitt è una traslocazione intercromosomica reciproca che coinvolge i cromosomi 8 e 14, determinando l’attivazione dell’oncogene c- Myc sito sul cromosoma 8. La traslocazione cromosomica che coinvolge Myc è un esempio di attivazione genica per traslocazione cromosomica senza modificazione della sequenza codificante. La traslocazione cromosomica che coinvolge Myc è un esempio di attivazione genica per traslocazione cromosomica senza modificazione della sequenza codificante.

44
LINFOMA di BURKITT LINFOMA di BURKITT 1) Spostamento del proto-oncogene c-Myc dal cromosoma 8 al 14; 2) Traslocazione di c-Myc nel locus della catena pesante delle immunoglobuline (IgH) sul cromosoma 14; CONSEGUENZE CONSEGUENZE Iperproduzione della proteina Myc, che causa la trasformazione neoplastica. Iperproduzione della proteina Myc, che causa la trasformazione neoplastica. L’espressione deregolata di Myc determina la permanenza in ciclo delle cellule B e conseguentemente la loro trasformazione in senso tumorale. L’espressione deregolata di Myc determina la permanenza in ciclo delle cellule B e conseguentemente la loro trasformazione in senso tumorale.

45
Individui affetti da linfoma di Burkitt

46
Patologia Patologia Il linfoma di Burkitt puo' esordire all'improvviso come una tumefazione delle ossa mascellari rapidamente evolutiva che determina ulcerazioni della mucosa sovrastante oppure con sudorazioni improvvise e molto abbondanti soprattutto notturne se colpisce l'intestino. Il linfoma di Burkitt puo' esordire all'improvviso come una tumefazione delle ossa mascellari rapidamente evolutiva che determina ulcerazioni della mucosa sovrastante oppure con sudorazioni improvvise e molto abbondanti soprattutto notturne se colpisce l'intestino. All'esame radiografico si osserva un'area radiotrasparente di osteolisi con margini frastagliati e riassorbimento radicolare delle radici dei denti contigui alla lesione osteoclastica. All'esame istologico il linfoma di Burkitt ha un aspetto tipico definito a cielo stellato, costituito da cellule macrofagiche che hanno fagocitato dei detriti cellulari. All'esame radiografico si osserva un'area radiotrasparente di osteolisi con margini frastagliati e riassorbimento radicolare delle radici dei denti contigui alla lesione osteoclastica. All'esame istologico il linfoma di Burkitt ha un aspetto tipico definito a cielo stellato, costituito da cellule macrofagiche che hanno fagocitato dei detriti cellulari.

47
Terapie Terapie Il trattamento elettivo di tale affezione patologica è la polichemioterapia ad alte dosi associata ad anticorpo monoclonale Ritubimax (terapia R-BFM 6 cicli ravvicinati A1-B1-C1, A2-B2-C2, durata totale circa 6 mesi). Il tasso di sopravvivenza è alto, intorno al 90%. Il trattamento elettivo di tale affezione patologica è la polichemioterapia ad alte dosi associata ad anticorpo monoclonale Ritubimax (terapia R-BFM 6 cicli ravvicinati A1-B1-C1, A2-B2-C2, durata totale circa 6 mesi). Il tasso di sopravvivenza è alto, intorno al 90%.
. Il tasso di sopravvivenza è alto, intorno al 90%..")
48
2) Leucemia mieloide cronica Incidenza : 1-2 persone ogni 100.000 abitanti; rara sotto i dieci anni. L’età media della diagnosi è 45-55 anni. Incidenza : 1-2 persone ogni 100.000 abitanti; rara sotto i dieci anni. L’età media della diagnosi è 45-55 anni. Patogenes i: Nella grande maggioranza dei casi, la LMC sembra sia dovuta alla traslocazione reciproca di segmenti di genoma tra i cromosomi 9 e 22, con formazione del cosiddetto cromosoma Philadelphia (Ph), corrispondente a un cromosoma 22 in cui si è creato il gene di fusione BCR-ABL. Questo gene codifica per una proteina (tirosin-chinasi) che rende "immortali" i blasti ed è quindi importante sia nella patogenesi della CML sia nella sua espressione clinica. Patogenes i: Nella grande maggioranza dei casi, la LMC sembra sia dovuta alla traslocazione reciproca di segmenti di genoma tra i cromosomi 9 e 22, con formazione del cosiddetto cromosoma Philadelphia (Ph), corrispondente a un cromosoma 22 in cui si è creato il gene di fusione BCR-ABL. Questo gene codifica per una proteina (tirosin-chinasi) che rende "immortali" i blasti ed è quindi importante sia nella patogenesi della CML sia nella sua espressione clinica.
, corrispondente a un cromosoma 22 in cui si è creato il gene di fusione BCR-ABL. Questo gene codifica per una proteina (tirosin-chinasi) che rende immortali i blasti ed è quindi importante sia nella patogenesi della CML sia nella sua espressione clinica. Patogenes i: Nella grande maggioranza dei casi, la LMC sembra sia dovuta alla traslocazione reciproca di segmenti di genoma tra i cromosomi 9 e 22, con formazione del cosiddetto cromosoma Philadelphia (Ph), corrispondente a un cromosoma 22 in cui si è creato il gene di fusione BCR-ABL. Questo gene codifica per una proteina (tirosin-chinasi) che rende immortali i blasti ed è quindi importante sia nella patogenesi della CML sia nella sua espressione clinica..")
49
In figura è indicato il CROMOSOMA PHILADELPHIA responsabile della leucemia.

51
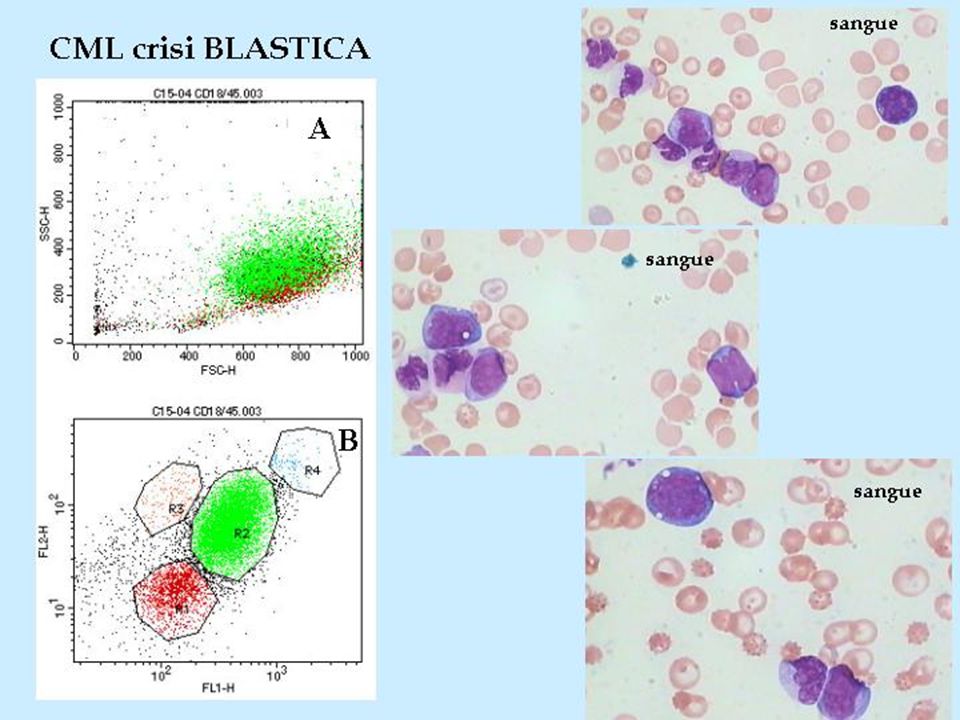
Fenotipo e storia naturale : Fenotipo e storia naturale : A causare la LMC è la moltiplicazione anomala di cellule staminali emopoietiche pluripotenti, cioè ancora in grado di proliferare e di differenziarsi. A causare la LMC è la moltiplicazione anomala di cellule staminali emopoietiche pluripotenti, cioè ancora in grado di proliferare e di differenziarsi. Le tre fasi della CML : Le tre fasi della CML : 1) La durata media della prima fase è di tre anni. Presenta una situazione clinica stabile e assenza o rarità di sintomi. 2) La seconda fase è caratterizzata da un netto peggioramento di tutti i parametri clinici ed ematologici. Dura pochi mesi. 3) La terza fase detta comunemente blastica, in assenza di terapia, la sopravvivenza è di poche settimane, ma anche nei malati sottoposti a trattamenti aggressivi la sopravvivenza media è di pochi mesi.
 La durata media della prima fase è di tre anni. Presenta una situazione clinica stabile e assenza o rarità di sintomi. 2) La seconda fase è caratterizzata da un netto peggioramento di tutti i parametri clinici ed ematologici. Dura pochi mesi. 3) La terza fase detta comunemente blastica, in assenza di terapia, la sopravvivenza è di poche settimane, ma anche nei malati sottoposti a trattamenti aggressivi la sopravvivenza media è di pochi mesi..")
53
Trattamento: Il trapianto allogenico di midollo (BMT) è la via più comunemente intrapresa. I pazienti che non posso effettuare il BMT vengo trattati con interferone. L’ultima scoperta nell’ambito dei farmaci molecolari è “L’Imatinib”, ossia un inibitore delle tirosin-chinasi BCR-ABL, che è risultato ben tollerato. Trattamento: Il trapianto allogenico di midollo (BMT) è la via più comunemente intrapresa. I pazienti che non posso effettuare il BMT vengo trattati con interferone. L’ultima scoperta nell’ambito dei farmaci molecolari è “L’Imatinib”, ossia un inibitore delle tirosin-chinasi BCR-ABL, che è risultato ben tollerato.
 è la via più comunemente intrapresa. I pazienti che non posso effettuare il BMT vengo trattati con interferone. L’ultima scoperta nell’ambito dei farmaci molecolari è L’Imatinib , ossia un inibitore delle tirosin-chinasi BCR-ABL, che è risultato ben tollerato..")
54
3) Sindrome di Down familiare La forma familiare della sindrome di Down è dovuta ad una traslocazione che vede coinvolti i cromosomi 14 (o 15) e 21. La forma familiare della sindrome di Down è dovuta ad una traslocazione che vede coinvolti i cromosomi 14 (o 15) e 21. Gli individui affetti,pur presentando un normale assetto cromosomico, hanno una PARZIALE TRISOMIA del cromosoma 21. Gli individui affetti,pur presentando un normale assetto cromosomico, hanno una PARZIALE TRISOMIA del cromosoma 21. Cromosoma traslocato ereditato da uno dei genitori Cromosoma traslocato ereditato da uno dei genitori
 e 21. Gli individui affetti,pur presentando un normale assetto cromosomico, hanno una PARZIALE TRISOMIA del cromosoma 21. Gli individui affetti,pur presentando un normale assetto cromosomico, hanno una PARZIALE TRISOMIA del cromosoma 21. Cromosoma traslocato ereditato da uno dei genitori Cromosoma traslocato ereditato da uno dei genitori.")
55
Individui affetti da sindrome di Down

56
Responsabile della sindrome è una traslocazione robertsoniana Responsabile della sindrome è una traslocazione robertsoniana (o fusione centrica): (o fusione centrica): Si forma un singolo cromosoma submetacentrico grazie alla fusione dei bracci lunghi dei cromosomi 14 e 21. Si forma un singolo cromosoma submetacentrico grazie alla fusione dei bracci lunghi dei cromosomi 14 e 21.

57
Un soggetto portatore della traslocazione alla conta dei cromosomi presenta 45 cromosomi, ma ad un’accurata analisi citogenetica in esso si rileva una compiuta diploidia (soggetto FALSO MONOSOMICO) Un soggetto portatore della traslocazione alla conta dei cromosomi presenta 45 cromosomi, ma ad un’accurata analisi citogenetica in esso si rileva una compiuta diploidia (soggetto FALSO MONOSOMICO) Un tale soggetto non presenta sintomi fenotipici della malattia, ma, avendo un cromosoma traslocato, può generare figli affetti dalla sindrome di Down. Un tale soggetto non presenta sintomi fenotipici della malattia, ma, avendo un cromosoma traslocato, può generare figli affetti dalla sindrome di Down.

58
Traslocazione 21/14 (robertsoniana) Possibili effetti della traslocazione robertsoniana 21/14
 Possibili effetti della traslocazione robertsoniana 21/14")
Presentazioni simili















>")






