Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Sep, 14, 2010 Chieti,.....” E quale è quei che cade, e non sa como, per forza di demon ch’a terra il tira........” “La Divina Commedia” (1302-1321), Inferno, Canto XXIV, 112-117 Le Epilessie Idiopatiche
, Inferno, Canto XXIV, Le Epilessie Idiopatiche")
2
Epilessia dal greco " Epilambanein " " Colto di sorpresa "

3
John Huglings-Jackson definisce l’epilessia come noi la conosciamo (1873) “Epilepsy is the name for occasional, sudden, excessive, rapid, and local discharges of grey matter.” “L’epilessia è la denominazione di una scarica occasionale, improvvisa, rapida e localizzata della sostanza grigia.” _______________________________ John Huglings-Jackson (1835-1911) era medico del National Hospital e del London Hospital di Londra
 Epilepsy is the name for occasional, sudden, excessive, rapid, and local discharges of grey matter. L’epilessia è la denominazione di una scarica occasionale, improvvisa, rapida e localizzata della sostanza grigia. _______________________________ John Huglings-Jackson ( ) era medico del National Hospital e del London Hospital di Londra")
4
CRISI EPILETTICA Disturbo “parossistico” ( a inizio e fine improvvisi) dovuto a una scarica neuronale abnorme nei circuiti di una parte o di tutto il cervello
 dovuto a una scarica neuronale abnorme nei circuiti di una parte o di tutto il cervello")
5
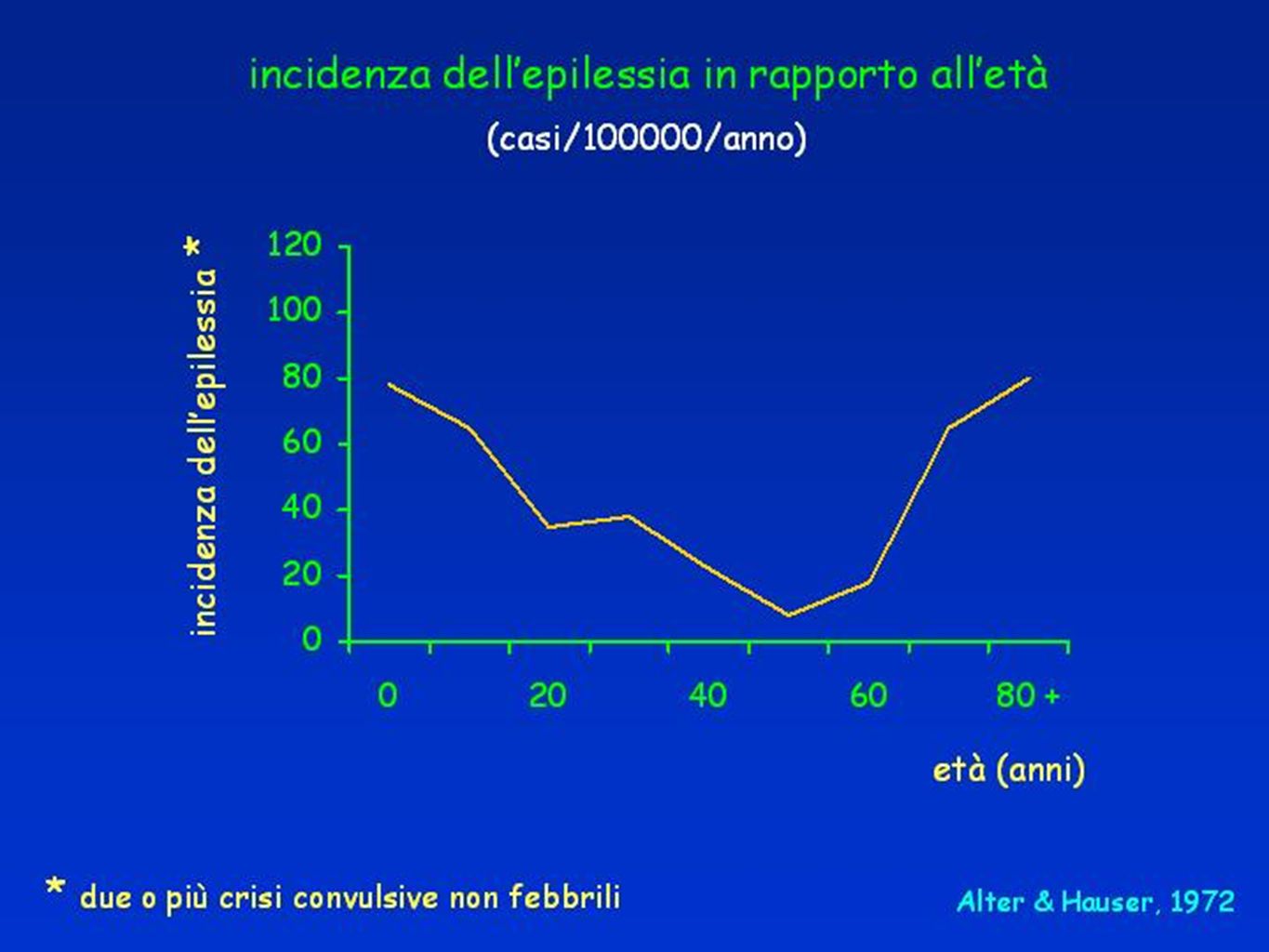
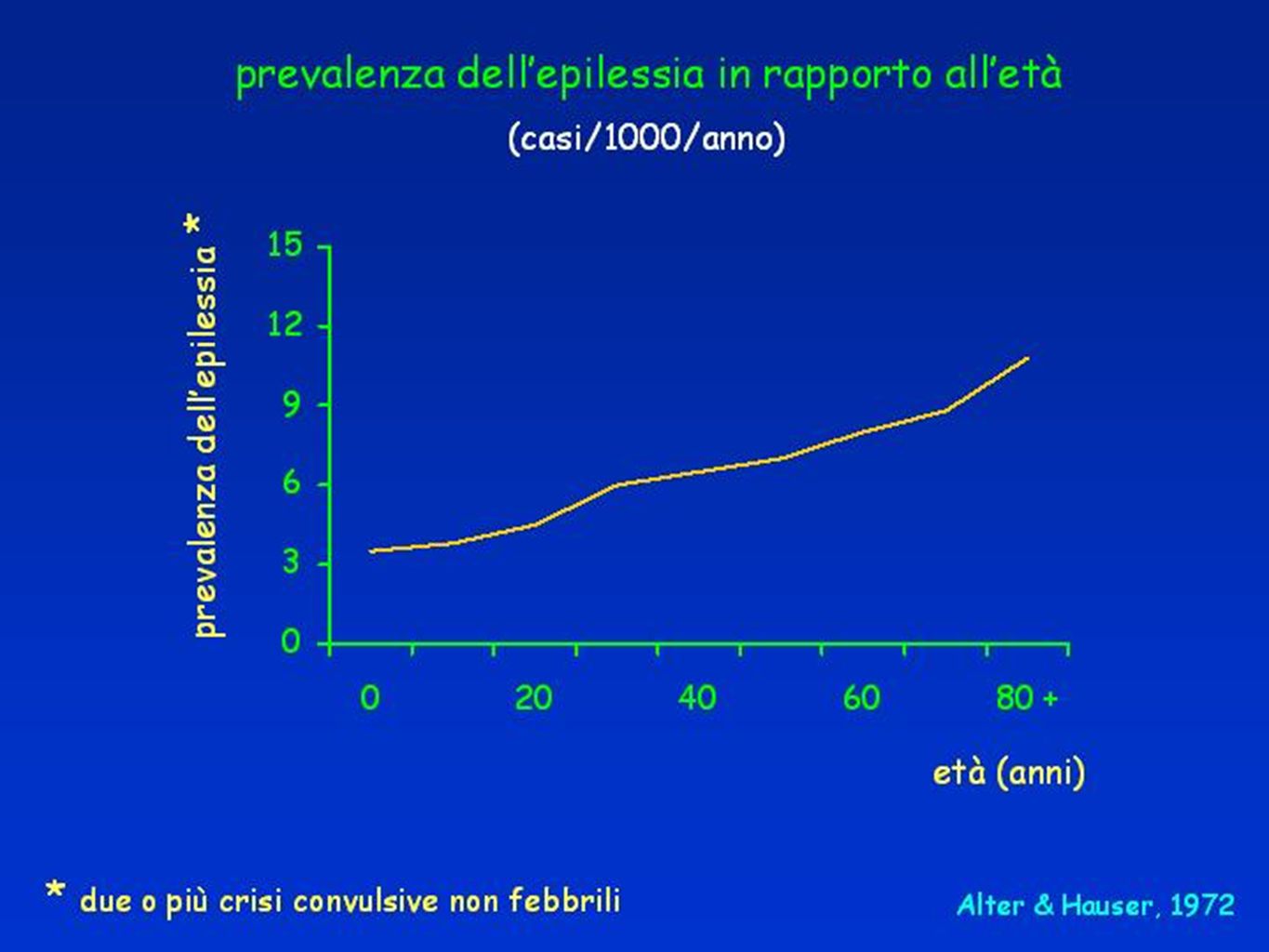
PREVALENZA CRISI EPILETTICA OCCASIONALE NELLA POPOLAZIONE GENERALE Stima OMS

6
EPILESSIE Prevalenza 0,5 - 2 % abitanti Incidenza 20-70 casi/100.000 50 milioni di pazienti nel mondo.

10
Perché si configuri un’ Epilessia o una Sindrome Epilettica sono necessari altri 2 elementi : - la ricorrenza delle crisi ( epilessia - la ricorrenza delle crisi (La diagnosi di epilessia è formulata dopo il verificarsi di almeno due eventi critici). - la spontaneità delle crisi (le crisi non devono essere provocate da situazioni particolari come febbre o da patologie acute del sistema nervoso centrale, come encefaliti, squilibri metabolici acuti, abuso di alcol o di droghe o da un trauma cranico nell’immediato o da un accidente vascolare nelle prime ore o nei primi giorni) Queste crisi verranno definite più opportunamente crisi epilettiche occasionali o crisi sintomatiche acute. Queste crisi verranno definite più opportunamente crisi epilettiche occasionali o crisi sintomatiche acute. Epilessia e sindromi epilettiche
 Queste crisi verranno definite più opportunamente crisi epilettiche occasionali o crisi sintomatiche acute. Queste crisi verranno definite più opportunamente crisi epilettiche occasionali o crisi sintomatiche acute. Epilessia e sindromi epilettiche.")
11
Una crisi epilettica è definita da due elementi:Elemento clinico (manifestazione clinica accessuale)Elemento fisiopatologico (derivante da una particolare modalità di disfunzione dell'attività cerebrale, la scarica parossistica). Crisi epilettiche

12
Epilessia e sindromi epilettiche Definizione e criteri diagnostici Definizione e criteri diagnostici Si usano due differenti tipi di classificazioni: -Classificazione delle crisi epilettiche -Classificazione delle sindromi epilettiche ILAE L’inquadramento del tipo di crisi e di epilessia o della sindrome epilettica è in accordo alle odierne classificazioni (ILAE).
.")
13
La classificazione internazionale delle crisi epilettiche del La classificazione internazionale delle crisi epilettiche del distingue: crisi focali crisi focali (già parziali), la cui manifestazione critica coinvolge una determinata area cerebrale e la cui scarica epilettica può rimanere localizzata o diffondere alle aree circostanti; crisi generalizzate crisi generalizzate (convulsive o non convulsive), le cui manifestazioni critiche coinvolgono sincronicamente e simmetricamente i due emisferi. Le crisi epilettiche

14
CRISI FOCALI CRISI FOCALI SEMPLICICRISI FOCALI SEMPLICI CRISI FOCALI COMPLESSE (con alterazione dello stato di coscienza)CRISI FOCALI COMPLESSE (con alterazione dello stato di coscienza) LE CRISI FOCALI POSSONO SECONDARIAMENTE GENERALIZZARELE CRISI FOCALI POSSONO SECONDARIAMENTE GENERALIZZARE TIPI DI CRISI FOCALI TIPI DI CRISI FOCALI Motorie (cloniche, toniche, miocloniche) Sensitive Sensoriali (olfattive, gustative, visive …) Vegetative Affettive (paura, angoscia….) Discognitive (afasiche; dismnesiche; deja vu; deja vecu …) Etc…
CRISI FOCALI COMPLESSE (con alterazione dello stato di coscienza) LE CRISI FOCALI POSSONO SECONDARIAMENTE GENERALIZZARELE CRISI FOCALI POSSONO SECONDARIAMENTE GENERALIZZARE TIPI DI CRISI FOCALI TIPI DI CRISI FOCALI Motorie (cloniche, toniche, miocloniche) Sensitive Sensoriali (olfattive, gustative, visive …) Vegetative Affettive (paura, angoscia….) Discognitive (afasiche; dismnesiche; deja vu; deja vecu …) Etc…")
15
Le epilessie Semeiologia delle crisi Crisi generalizzate - assenze - mioclonie - crisi tonico-cloniche - crisi toniche - crisi atoniche - crisi cloniche

16
Assenze Sono crisi di breve durata, caratterizzate da alterazioni (attenuazione o sospensione) dello stato di coscienza. Si distinguono in: - Assenze Tipiche - Assenze Atipiche

18
Assenze tipiche Sono crisi con inizio e fine bruschi e si accompagnano ad una scarica bilaterale sincrona e simmetrica di punte-onda a 3-4 Hz, regolari e di grande ampiezza con attività di fondo normale. Le crisi sono pluriquotidiane e si distinguono 6 varietà cliniche: -assenze semplici (alterazione isolata della coscienza); -assenze con elementi clonici (clonie palpebrali o buccali a 3 c/s); -assenze con componente tonica (revulsione dei globi oculari o estensione della testa); -assenze con componente atonica (riduzione del tono con flessione della testa e, talvolta, del tronco); -assenze con automatismi gestuali semplici; -assenze con elementi vegetativi (modificazione della respirazione, midriasi, enuresi).
; -assenze con elementi clonici (clonie palpebrali o buccali a 3 c/s); -assenze con componente tonica (revulsione dei globi oculari o estensione della testa); -assenze con componente atonica (riduzione del tono con flessione della testa e, talvolta, del tronco); -assenze con automatismi gestuali semplici; -assenze con elementi vegetativi (modificazione della respirazione, midriasi, enuresi)..")
19
Assenze Atipiche Sono crisi con inizio e fine non bruschi, caratterizzate da scariche di punte-onda generalizzate a frequenza inferiore a 3 Hz. L’attività di fondo è spesso normale. Sono frequentemente presenti elementi atonici o tonici più intensi che nelle assenze tipiche. L’alterazione della coscienza è spesso meno profonda e di intensità variabile.

20
Crisi Miocloniche Sono caratterizzate da scosse muscolari brevi, a scatti, bilaterali e simmetriche, di topografia ed intensità variabili, che possono provocare caduta quando sono agli arti inferiori e particolarmente intense. Non vi sono alterazioni percettibili della coscienza. L’EEG mostra polipunte-onda bilaterali, sincrone con le scosse.

21
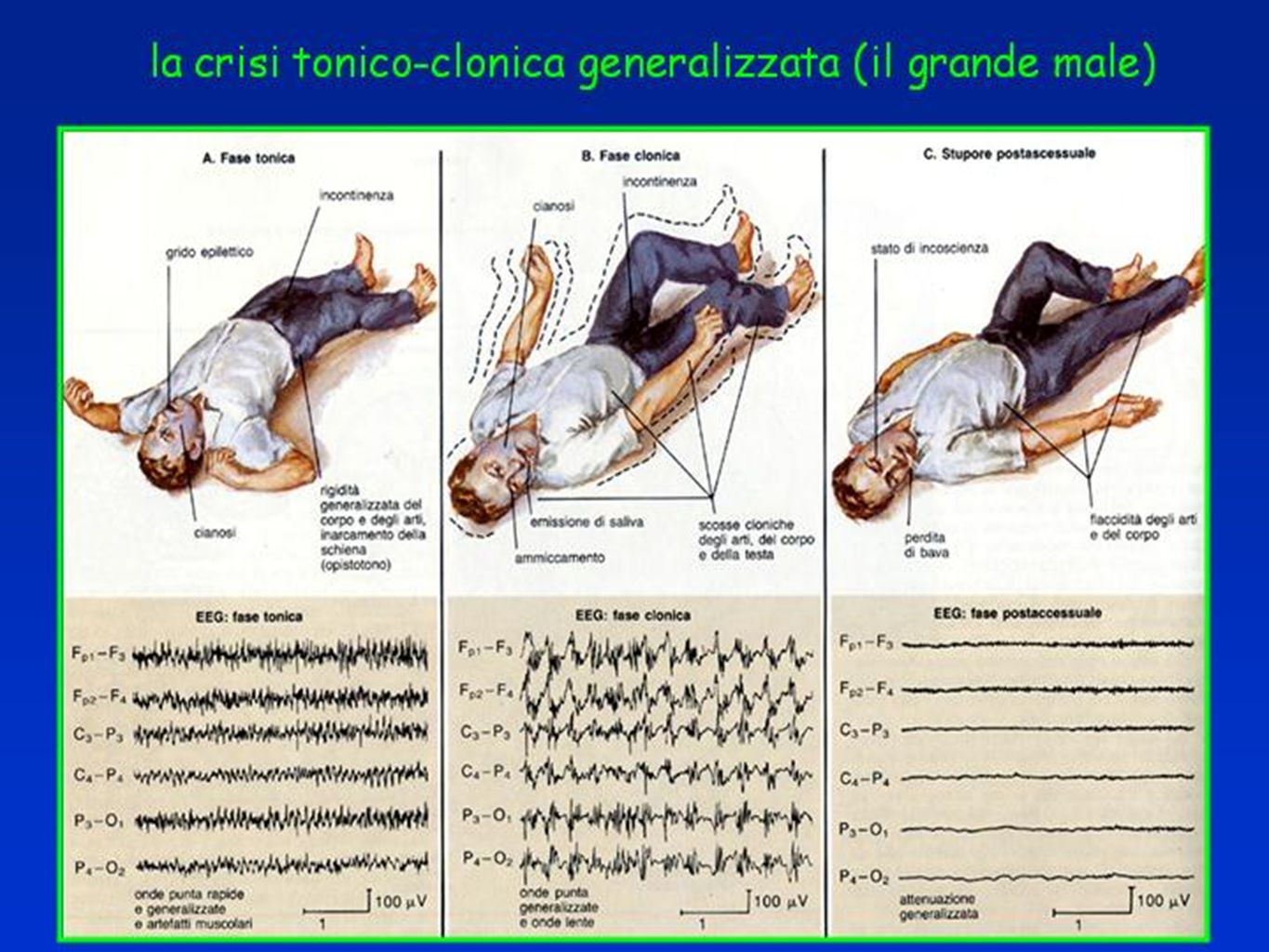
Crisi Tonico-cloniche 1) Fase tonica : abolizione iniziale della coscienza, contrazione tonica della muscolatura scheletrica della durata di 10-20 sec. Sono associate importanti turbe vegetative (tachicardia, aumento della pressione arteriosa, ipersecrezione bronchiale, arrossamento del volto midriasi…). La tetanizzazione progressivamente si frammenta conducendo alla fase clonica. 2) Fase clonica : dura circa 30 secondi, si esprime con scosse bilaterali che si distanziano progressivamente fino ad interrompersi bruscamente. Il volto è cianotico 3) Fase post-critica : dura da alcuni minuti ad alcune ore. Il soggetto è obnubilato, immobile, ipotonico. La respirazione riprende rumorosa ed il livello di coscienza migliora progressivamente. L’EEG critico attività rapida di basso voltaggio e di ampiezza crescente cui fanno seguito nella fase clonica, polipunte o polipunte-onde
. La tetanizzazione progressivamente si frammenta conducendo alla fase clonica. 2) Fase clonica : dura circa 30 secondi, si esprime con scosse bilaterali che si distanziano progressivamente fino ad interrompersi bruscamente. Il volto è cianotico 3) Fase post-critica : dura da alcuni minuti ad alcune ore. Il soggetto è obnubilato, immobile, ipotonico. La respirazione riprende rumorosa ed il livello di coscienza migliora progressivamente. L’EEG critico attività rapida di basso voltaggio e di ampiezza crescente cui fanno seguito nella fase clonica, polipunte o polipunte-onde.")
23
Crisi tonico-clonica (Grande Male)
")
24
CLASSIFICAZIONE DELLE EPILESSIE A SECONDA DELL’ EZIOLOGIA EPILESSIE IDIOPATICHEEPILESSIE IDIOPATICHE EPILESSIE CRIPTOGENICHEEPILESSIE CRIPTOGENICHE EPILESSIE SINTOMATICHEEPILESSIE SINTOMATICHE

25
CLASSIFICAZIONE DELLE EPILESSIE A SECONDA DELL’ EZIOLOGIA EPILESSIE IDIOPATICHE Causa presumibilmente genetica Generalmente con evoluzione benigna sintomatiche o secondarie Epilessie sintomatiche o secondarie Epilessie sostenute da una lesione strutturale del sistema nervoso centrale (SNC) criptogenetiche o probabilmente sintomatiche Epilessie criptogenetiche o probabilmente sintomatiche la natura sintomatica è soltanto sospettata sulla base delle caratteristiche cliniche, EEG ed evolutive, ma non è possibile dimostrarla con le indagini attualmente disponibili
 criptogenetiche o probabilmente sintomatiche Epilessie criptogenetiche o probabilmente sintomatiche la natura sintomatica è soltanto sospettata sulla base delle caratteristiche cliniche, EEG ed evolutive, ma non è possibile dimostrarla con le indagini attualmente disponibili")
26
Fattori che suggeriscono causa genetica... Concordanza nei gemelli: 50% in monozigoti; 10% in dizigoti (affected sib => risk factor 100) Tra coppie concordanti, >80% mostra stessa forma di epilessia Presenza di famiglia in cui segrega come tratto monogenico
 Tra coppie concordanti, >80% mostra stessa forma di epilessia Presenza di famiglia in cui segrega come tratto monogenico.")
27
Fisiopatologia delle Epilessie idiopatiche La crisi epilettica è conseguenza dell'attivazione eccessiva ed ipersincrona dei neuroni di un'area corticale di estensione variabile. L’eccitabilità della membrana dipende da scambi ionici che si verificano mediante differenti sistemi basati su meccanismi attivi (pompe ioniche), su diffusione attraverso canali ionici attivati chimicamente (neurotrasmettiotre-recettore) o dal livello di polarizzazione della membrana (voltaggio dipendenti). Anomalie che coinvolgono questi recettori e questi canali ionici sono alla base di alcune condizioni epilettiche geneticamente trasmesse
, su diffusione attraverso canali ionici attivati chimicamente (neurotrasmettiotre-recettore) o dal livello di polarizzazione della membrana (voltaggio dipendenti). Anomalie che coinvolgono questi recettori e questi canali ionici sono alla base di alcune condizioni epilettiche geneticamente trasmesse.")
29
Quando?

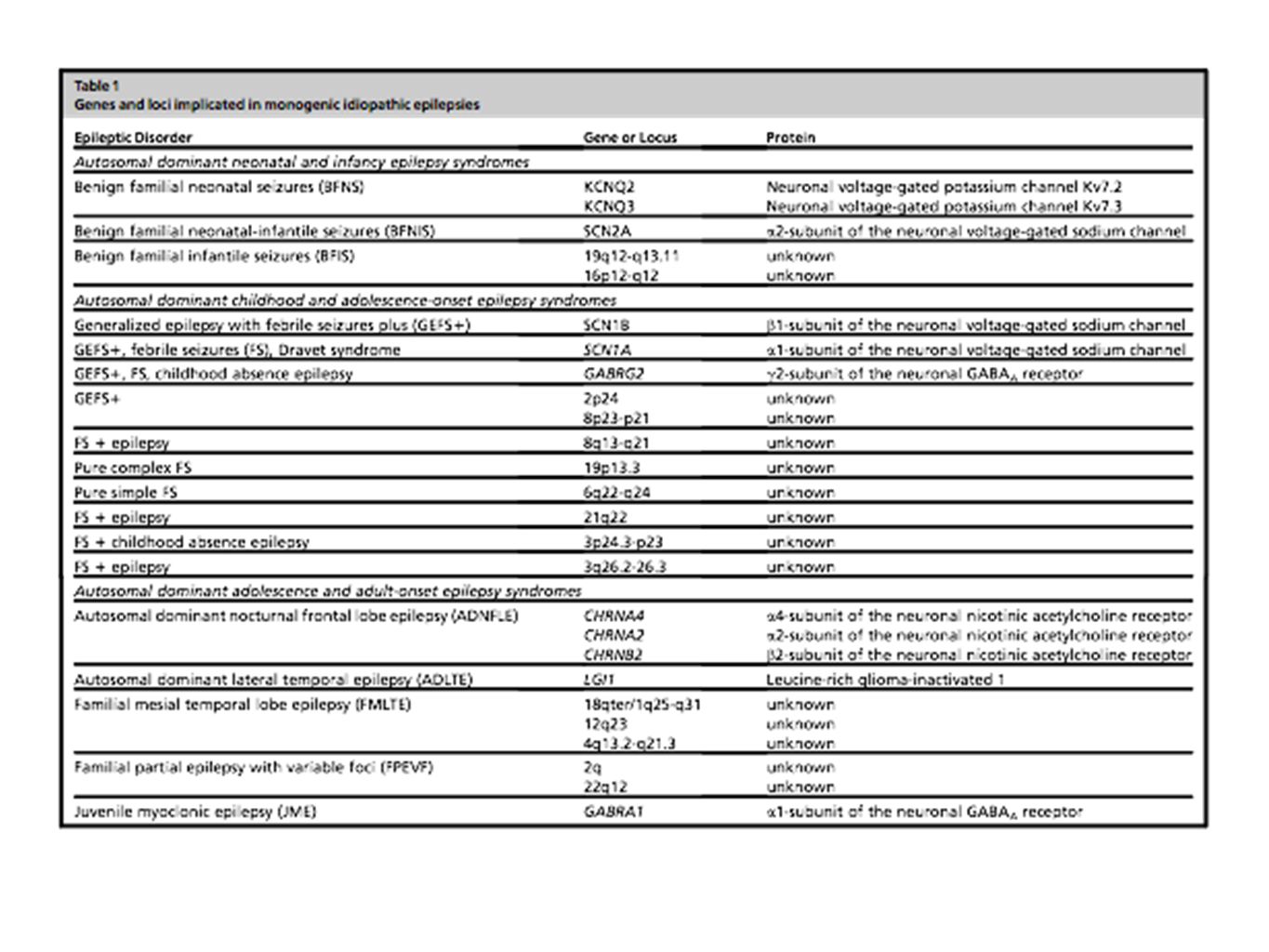
30
Benign Familial Neonatal Seizures BFNS di solito inizia il secondo o terzo giorno di vita e spesso cessa spontaneamente dopo alcune settimane o mesi. Si possono presentare varie manifestazioni tra cui gli attacchi tonici, apnea, e cloniche, focale, e le caratteristiche autonomica. La maggior parte cessano dopo 4 mesi, ma, a volte, convulsioni febbrili si verificano nel 5% e l'epilessia nel 11% degli individui. La BFNS è una epilessia focale autosomica dominante, in cui la stragrande maggioranza delle famiglie hanno mutazioni in KCNQ2, con pochi in KCNQ37 codificanti, rispettivamente,per le subunità del canale potassio voltaggio-dipendenti Kv7.2 e Kv7.3. Circa 70 mutazioni in KCNQ2 e 6 in KCNQ3 sono state identificate in famiglie colpite da questa sindrome epilessia (Human Gene Mutation Database). Mutazioni de novo in KCNQ2 sono stati riportati anche in pazienti sporadici con benigna neonatale seizures. Quasi la metà delle mutazioni KCNQ2 si pensa possano troncare la proteina e il resto sono mutazioni missense. Nel gene KCNQ3, solo mutazioni missense sono state identificate. Una piccola percentuale di famiglie non hanno mutazioni in nessuno dei due né KCNQ2 KCNQ3, sottolineando che almeno un terzo gene deve essere coinvolto
. Mutazioni de novo in KCNQ2 sono stati riportati anche in pazienti sporadici con benigna neonatale seizures. Quasi la metà delle mutazioni KCNQ2 si pensa possano troncare la proteina e il resto sono mutazioni missense. Nel gene KCNQ3, solo mutazioni missense sono state identificate. Una piccola percentuale di famiglie non hanno mutazioni in nessuno dei due né KCNQ2 KCNQ3, sottolineando che almeno un terzo gene deve essere coinvolto.")
31
Benign Familial Neonatal-infantile Seizures L'età di esordio all'interno di nuclei familiari con BFNIS è da 2 giorni a 7 mesi con la maggior parte dei casi con insorgenza intorno ai 2-3 mesi di età. La maggior parte delle famiglie con BFNIS hanno mutazioni in SCN2A, che codifica per la subunità a2 del canale neuronale sodio voltaggio-dipendenti. Ad oggi, 10 mutazioni missense nel gene SCN2A sono stati segnalati come causa di BFNIS. L'identificazione di BFNIS con mutazioni SCN2A ha così contribuito a delineare le sindromi epilessia benigna dei periodi neonatale e infantile. Il fenotipo clinico si sovrappone a BFNIS

32
Benign Familial Infantile Seizures BFIS è una sindrome ereditaria autosomica dominante di epilessia parziale della prima l'infanzia (a partire dai 4 agli 8 mesi) con remissione spontanea prima dell'età di 3 anni. Le crisi si presentano spesso come un gruppo in pochi giorni, sollevando preoccupazione per una condizione più grave. Esse hanno caratteristiche motorie e insorgenza focale visibile su un EEG. Due loci genetici sono stati segnalati in BFIS famiglie con trasmissione AD: uno sul cromosoma 19q12-q13.11 in cinque famiglie italiane e uno sul 16p12-q12. Nessun gene è stato ad oggi identificato.

33
Generalized Epilepsy with Febrile Seizures Plus L’Epilessia generalizzata con convulsioni febbrili plus (GEFS1) è una epilessia autosomica dominante riconosciuta come un'entità clinica distinta nel 1997 con caratteristico, ma eterogeneo, fenotipo clinico nei membri affetti. Sebbene GEFS1 sindrome presenti una grande eterogeneità fenotipica, la caraterizzazione clinica di GEFS1 è stato convalidato dall'identificazione dei singoli geni principali: SCN1A, SCN1B e GABRG2. (solo il 15%) SCN1B : Mutazione missense C121W in una famiglia Australiana; altre due mutazioni (R85C and R85H) in due nuove famiglie australiane. SCN1A: Due mutazioni son state identificate in fue famiglie francesi (Thr875Met and Arg1648His). Da allora (2000) più di 20 mutazioni son state associate con fenotipi GEFS+. Vista la presenza di convulsioni febbrili in una forma di epilessia mioclonica infantile (SMEI, Dravet Syndrome),lo screening di questo gene ha portato all’individuazione di mutazioni in 7 bambini. GABRG2: Una mutazione missenso (K289M) è stata ritrovata in una famiglia con linkage nel cromosoma 5q14. UN altra mutazione missense è stata individuata missense mutation (R43Q) in un pedigree Australiano.
 SCN1B : Mutazione missense C121W in una famiglia Australiana; altre due mutazioni (R85C and R85H) in due nuove famiglie australiane. SCN1A: Due mutazioni son state identificate in fue famiglie francesi (Thr875Met and Arg1648His). Da allora (2000) più di 20 mutazioni son state associate con fenotipi GEFS+. Vista la presenza di convulsioni febbrili in una forma di epilessia mioclonica infantile (SMEI, Dravet Syndrome),lo screening di questo gene ha portato all’individuazione di mutazioni in 7 bambini. GABRG2: Una mutazione missenso (K289M) è stata ritrovata in una famiglia con linkage nel cromosoma 5q14. UN altra mutazione missense è stata individuata missense mutation (R43Q) in un pedigree Australiano..")
34
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy Esordio prime 2 decadi Clusters di crisi motorie notturne, con manifestazioni che emergono dal lobo frontale prevalentemente nel sonno. Penetranza incompleta CHRNA4 (20 q) (Ser248Phe) in a large Australian kindred CHRNB2 due mutazioni in una famiglia italiana e una scozzese (V287L-V287M) CHRNA2 una mutazione in una famiglia italiana I279N
 (Ser248Phe) in a large Australian kindred CHRNB2 due mutazioni in una famiglia italiana e una scozzese (V287L-V287M) CHRNA2 una mutazione in una famiglia italiana I279N.")
35
Autosomal Dominant Lateral Temporal Lobe Epilepsy con sintomi uditivi Sintomi focali visivi, afasici, pseudovertiginosi, con o senza generalizzazione. La componente uditiva, caratteristica, si presentava come un rumore di frigo, radio o dispercezione dell’ambiete circostante. Un locus è stato mappato nel cromosoma e mutazioni son state individuate nel gene LGI1 (leucine-rich glioma-inactivated 1) che oggi è mutato in circa il 50% delle famiglie con tale forma di epilessia. Juvenile Myoclonic Epilepsy 3-12% di tutte le forme erediarie. Esordisce nell’adolescenza con crisi tonico-cloniche, mioclono, assenze. Presenta alterazioni EEGrafiche con polipunta onda e frequenza di 3- 6Hz. Locus chr 5q34. Una mutazione (A322D) è stata identificata nel gene che codifica l subunità A1 del recettore GABAA.
 che oggi è mutato in circa il 50% delle famiglie con tale forma di epilessia. Juvenile Myoclonic Epilepsy 3-12% di tutte le forme erediarie. Esordisce nell’adolescenza con crisi tonico-cloniche, mioclono, assenze. Presenta alterazioni EEGrafiche con polipunta onda e frequenza di 3- 6Hz. Locus chr 5q34. Una mutazione (A322D) è stata identificata nel gene che codifica l subunità A1 del recettore GABAA..")
36
Epilessia e sindromi epilettiche Definizione e criteri diagnostici Definizione e criteri diagnostici Si usano due differenti tipi di classificazioni: -Classificazione delle crisi epilettiche -Classificazione delle sindromi epilettiche Sindrome epilettica idiopatica: una sindrome che è solo epilessia senza lesioni strutturali o altri segni o sintomi neurologici. Si presume sia su base genetica ed è usualmente età-dipendente (termine invariato). Idiopatico non è sinonimo di benigno.
. Idiopatico non è sinonimo di benigno..")
37
Classificazione internazionale delle epilessie e delle sindromi epilettiche. Epilessia e sindromi epilettiche parziali Idiopatiche Idiopatiche (con esordio legato all’ età) Epilessia benigna dell’ infanzia con punte centro-temporali Epilessia benigna dell’ infanzia con punte occipitali Tipo Panayotopulos (ad esordio precoce) Tipo Gastaut (ad esordio tardivo)
 Epilessia benigna dell’ infanzia con punte centro-temporali Epilessia benigna dell’ infanzia con punte occipitali Tipo Panayotopulos (ad esordio precoce) Tipo Gastaut (ad esordio tardivo).")
38
Epilessia parziale benigna a punte centro-temporali(epilessia rolandica di Loiseau) Esordio: 3-13 anni Crisi parziali semplici emifacciali o brachio-facciali, talora con secondaria generalizzazione (più frequente nel bambino piccolo) Segni tipici: parestesie al cavo orale, arresto della parola. Spesso notturne. EEG intercritico estremamente evocatore: punte lente di ampio voltaggio in regione centro-temporale spettacolarmente attivate dal sonno.

39
Epilessia a parossismi occipitali: Tipo Panayotopulos (ad esordio precoce) Esordio a 2-8 anni, remissione prima dei 12 aa. Prevalenza nel sesso femminile. Crisi prevalentemente notturne, spesso prolungate(anche ore), con pallore, nausea, vomito, sensazione di malessere, deviazione tonica degli occhi, talora emiconvulsione o crisi tonico-clonica generalizzata. Coscienza e linguaggio conservati in fase iniziale. Talora “sincope ictale”: il bambino diventa flaccido e non responsivo, prima della convulsione o come unico sintomo. EEG: Punte, punte-onda e onde lente angolari, multifocali, prevalentemente occipitali, ma anche rolandiche e frontali, attivate dal sonno, su una attività di fondo normale.
, con pallore, nausea, vomito, sensazione di malessere, deviazione tonica degli occhi, talora emiconvulsione o crisi tonico-clonica generalizzata. Coscienza e linguaggio conservati in fase iniziale. Talora sincope ictale : il bambino diventa flaccido e non responsivo, prima della convulsione o come unico sintomo. EEG: Punte, punte-onda e onde lente angolari, multifocali, prevalentemente occipitali, ma anche rolandiche e frontali, attivate dal sonno, su una attività di fondo normale..")
40
Epilessia a parossismi occipitali: Tipo Gastaut (ad esordio tardivo) Esordio a 3-15 anni, senza prevalenza di sesso. Crisi visive, con allucinazioni semplici o con amaurosi transitoria, che talora divengono parziali complesse o si generalizzano. Spesso deviazione del capo e degli occhi. Ricorrenza diurna delle crisi. Frequente cefalea post-critica, con caratteristiche di emicrania. EEG: parossismi occipitali, spesso bilaterali e sincroni, che scompaiono con la eliminazione della visione centrale (buio, chiusura degli occhi, lenti speciali). Prognosi meno buona rispetto alla forma di Panayotopulos
. Prognosi meno buona rispetto alla forma di Panayotopulos.")
41
ALTRE EPILESSIE PARZIALI BENIGNE DELL’INFANZIA - Epilessia occipitale idiopatica fotosensibile -Epilessia con crisi a semiologia affettiva -Epilessia benigna dell’infanzia con punte parietali e frequenti potenziali evocati somatosensitivi giganti -Epilessia benigna dell’infanzia con punte frontali e centrali. -Epilessia benigna dell’infanzia con punte-onda centrali e al vertice solo in sonno lento

42
Epilessie familiari focali autosomiche dominanti Epilessia familiare temporale mesiale trasmissione autosomica dominante a penetranza incompleta(60%). Esordio nell’adolescenza o nel giovane adulto. Prevale nel sesso femminile. Le crisi sono nel 90% dei casi parziali semplici con déjà vu, altri fenomeni esperenziali, paura, panico e allucinazioni, talora associati a segni vegetativi. Buona risposta alla terapia. Molti pazienti con crisi lievi e rare vengono diagnosticati solo per la presenza in famiglia di casi ad espressione clinica più chiara. Epilessia familiare temporale laterale(LFTLE) gene LGI1(leucine-rich, glioma inactivated 1 gene), anche detto Epitempina. E’ stato per la prima volta descritto nei tumori gliali nei quali può essere deleto o riarrangiato, ma nei quali non vi è una elevata frequenza di LFTLE. Tale gene può influenzare la migrazione neuronale o l’organizzazione corticale. Le mutazioni del LGI1 provocano rottura di proteine. Le mutazioni del LGI1 sono specifiche per LFTLE, ma non si ritrovano in tutte le famiglie affette. Esordio nell’adolescenza o giovane adulto. Crisi lievi e rare, con allucinazioni uditive Crisi di GM prevalentemente notturne e infrequenti. Ottima risposta alla terapia.
 gene LGI1(leucine-rich, glioma inactivated 1 gene), anche detto Epitempina. E’ stato per la prima volta descritto nei tumori gliali nei quali può essere deleto o riarrangiato, ma nei quali non vi è una elevata frequenza di LFTLE. Tale gene può influenzare la migrazione neuronale o l’organizzazione corticale. Le mutazioni del LGI1 provocano rottura di proteine. Le mutazioni del LGI1 sono specifiche per LFTLE, ma non si ritrovano in tutte le famiglie affette. Esordio nell’adolescenza o giovane adulto. Crisi lievi e rare, con allucinazioni uditive Crisi di GM prevalentemente notturne e infrequenti. Ottima risposta alla terapia..")
43
Convulsioni familiari neonatali benigne Convulsioni neonatali benigne Epilessia mioclonica benigna dell’ infanzia Piccolo-male-assenza dell’ infanzia Piccolo-male-assenza dell’adolescenza Epilessia mioclonica giovanile Epilessia con crisi tonico cloniche al risveglio Altre epilessie generalizzate non definite sopra Epilessie generalizzate con crisi scatenate da specifiche modalità di attivazione Classificazione internazionale delle epilessie e delle sindromi epilettiche. Epilessie e Sindromi epilettiche generalizzate Idiopatiche Epilessie generalizzate idiopatiche Età dipendenti Frequente familiarità per epilessia Crisi generalizzate Normalità neurologica e intellettiva Neuroimaging normale Punte e polipunte-onda generalizzate a 3 hertz o più. Attività di fondo normale Buona risposta alla terapia

44
Epilessia assenze dell’infanzia(Piccolo male) Età media di esordio a 6-7 anni Assenze tipiche, semplici o con automatismi, Tipicamente scatenate dall’iperpnea Punte-onda a 3 Hertz generalizzate Ottima risposta a etosuccimide o valproato Nel 40% dei casi insorgenza di crisi di grande male nell’adolescenza Epilessia assenze dell’adolescente Esordio più tardivo Le assenze sono più rare Le scariche di PO possono essere più rapide, a 4-5 hertz. Spesso si associano crisi di grande male Epilessia mioclonica giovanile benigna (Sindrome di Janz, Piccolo male mioclonico) Età di esordio 6-26 anni. Picco alla pubertà. Scosse miocloniche bilaterali, spesso simmetriche, isolate o ripetitive, tipicamente al risveglio e favorite dalla perdita di sonno. Rare crisi di grande male tipicamente precedute da mioclonie massive bilaterali(crisi clonico-tonico-cloniche) EEG critico e intercritico: PO rapide generalizzate Spesso fotosensibilità Forma geneticamente determinata Buona risposta al valproato Farmacodipendenza: recidiva delle crisi nel 90% dei casi dopo sospensione della terapia Epilessia con crisi di grande male al risveglio Esordio nell’adolescenza Crisi di grande male soprattutto dopo il risveglio mattutino Più frequente nel sesso femminile Fattori scatenanti: deprivazione di sonno, eccesso di alcool, brusco risveglio Talora fotosensibilità EEG intercritico: PO rapide generalizzate
 Età di esordio 6-26 anni. Picco alla pubertà. Scosse miocloniche bilaterali, spesso simmetriche, isolate o ripetitive, tipicamente al risveglio e favorite dalla perdita di sonno. Rare crisi di grande male tipicamente precedute da mioclonie massive bilaterali(crisi clonico-tonico-cloniche) EEG critico e intercritico: PO rapide generalizzate Spesso fotosensibilità Forma geneticamente determinata Buona risposta al valproato Farmacodipendenza: recidiva delle crisi nel 90% dei casi dopo sospensione della terapia Epilessia con crisi di grande male al risveglio Esordio nell’adolescenza Crisi di grande male soprattutto dopo il risveglio mattutino Più frequente nel sesso femminile Fattori scatenanti: deprivazione di sonno, eccesso di alcool, brusco risveglio Talora fotosensibilità EEG intercritico: PO rapide generalizzate.")
45
Mutazioni dei geni codificanti subunità del canale del Na voltaggio dipendente SCN1A Generalized Epilepsy & Febrile Seizures Plus (GEFS+) type 2 Severe Myoclonic Epilepsy of Infancy (SMEI) SCN1B GEFS+ type 1 SCN2A1 GEFS+ Benign Familial Neonatal- Infantile Seizures (BFNIS) Mutazioni dei geni codificanti subunità del canale del Cl voltaggio dipendente CLCN2A Juvenile Absence Epilepsy (JAE) Juvenile Myoclonic Epilepsy (JME) Epilepsy with Grand Mal upon Awakening (EGMA) Riepilogando........
 type 2 Severe Myoclonic Epilepsy of Infancy (SMEI) SCN1B GEFS+ type 1 SCN2A1 GEFS+ Benign Familial Neonatal- Infantile Seizures (BFNIS) Mutazioni dei geni codificanti subunità del canale del Cl voltaggio dipendente CLCN2A Juvenile Absence Epilepsy (JAE) Juvenile Myoclonic Epilepsy (JME) Epilepsy with Grand Mal upon Awakening (EGMA) Riepilogando")
46
Mutazioni dei geni codificanti subunità del canale del K voltaggio dipendente KCNQ2, KCNQ3 Benign Familial Neonatal Seizures (BFNS) KCND2 Temporal Lobe Epilepsy (TLE) KCNMA1 Generalized Epilepsy with Paroxysmal Dyskinesia (GEPD) Mutazioni dei geni codificanti subunità di recettori di neurotrasmettitori GABRG2 (GABA-receptor gamma-2 subunit) GEFS+ type 3 GABRA1 (GABA-receptor alpha-1 subunit) JME CHRNA4 (nicotinic acetylcholine receptor alpha-4 subunit) Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE) type 1 CHRNB2 (nicotinic acetylcholine receptor beta-2 subunit) CHRNa2 (nicotinic acetylcholine receptor alpha-2 subunit)
 KCND2 Temporal Lobe Epilepsy (TLE) KCNMA1 Generalized Epilepsy with Paroxysmal Dyskinesia (GEPD) Mutazioni dei geni codificanti subunità di recettori di neurotrasmettitori GABRG2 (GABA-receptor gamma-2 subunit) GEFS+ type 3 GABRA1 (GABA-receptor alpha-1 subunit) JME CHRNA4 (nicotinic acetylcholine receptor alpha-4 subunit) Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE) type 1 CHRNB2 (nicotinic acetylcholine receptor beta-2 subunit) CHRNa2 (nicotinic acetylcholine receptor alpha-2 subunit)")
47
Terapia Generalizzate: Valproato prima scelta anche se la lamotrigina, il levetiracetam e il topiramato Parziali: CBZ- lamtotrigina-Oxcarbazepina

Presentazioni simili



>")
, sta ad indicare una modalità di reazione.>")









>")





 da fattori potenzialmente dannosi per le cellule. Le cellule “offese” liberano molecole che.>")
>")
