Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Mechanisms of Disease Cardiac Plasticity Joseph A. Hill, M.D., Ph.D., and Eric N. Olson, Ph.D. N Engl J Med 2008;358: Condizioni che provocano il rimodellamento del cuore e ne causano atrofia o ipertrofia. A seconda dei casi il rimodel-lamento può essere normale o patologico. Il rimodellamento patologico si associa alla tendenza allo scompenso, alla dilatazione ventricolare, alla disfunzione sistolica e a modificazioni elettrofisiologiche che provocano aritmie ventricolari maligne.

2
Limiti dell’atrofia e dell’ipertrofia cardiaca: il cuore può andare incontro ad aumento o riduzione considerevole del volume, con un ambito dinamico del 100%

3
L’ipertrofia di un organo è caratterizzata da un aumento delle dimensioni delle cellule. Nell’accrescimento del cuore provocato dall’esercizio o dalla gravidanza, la struttura e la funzione sono normali e non vi è associazione con l’insufficienza cardiaca. Nell’ipertrofia che si verifica nell’ipertensione, nell’obesità, nelle malattie valvolari, dopo un infarto o in seguito ad alterazioni geniche delle proteine contrattili, vi sono alla base alterazioni metaboliche, strutturali e funzionali: il metabolismo vira verso la glicolisi (normalmente prevale il consumo di acidi grassi), i sarcomeri si disorganizzano, le correnti di calcio sono alterate, si riduce la contrattilità, si perdono miociti che vengono sostituiti da fibroblasti, compare disfunzione sistolica e/o diastolica e un “rimodellamento elettrico” (es. alterazioni dell’espressione e/o della funzione delle pompe ioniche). Tutto ciò rappresenta una serie di condizioni derivanti dalla plasticità miocardica.
, i sarcomeri si disorganizzano, le correnti di calcio sono alterate, si riduce la contrattilità, si perdono miociti che vengono sostituiti da fibroblasti, compare disfunzione sistolica e/o diastolica e un rimodellamento elettrico (es. alterazioni dell’espressione e/o della funzione delle pompe ioniche). Tutto ciò rappresenta una serie di condizioni derivanti dalla plasticità miocardica.")
4
L’accrescimento del cuore segue, classicamente, tre modelli morfologici:
Rimodellamento concentrico: aumento dello spessore (relativo) delle pareti senza aumento della massa; Ipertrofia concentrica: aumento dello spessore (relativo) e della massa con scarso aumento di volume; aggiunta di sarcomeri in parallelo e accrescimento laterale delle cellule: causa frequente l’ipertensione arteriosa e l’esercizio isometrico; Ipertrofia eccentrica: aumento della massa e del volume delle camere; lo spessore (relativo) può essere normale, aumentato o diminuito: cause esercizio isotonico, sovraccarico di volume, perdita di tessuto funzionante per infarto; aggiunta di sarcomeri in serie e allungamento dei miocardiociti. Fra gli atleti, quelli che hanno il cuore più grande sono i rematori, i ciclisti e gli sciatori di fondo; lo spessore di parete in genere non supera 1,3 cm (limite superiore per sedentari 1,1 cm). Gli allenamenti stagionali provocano variazioni stagionali delle dimensioni del cuore.
 delle pareti senza aumento della massa; Ipertrofia concentrica: aumento dello spessore (relativo) e della massa con scarso aumento di volume; aggiunta di sarcomeri in parallelo e accrescimento laterale delle cellule: causa frequente l’ipertensione arteriosa e l’esercizio isometrico; Ipertrofia eccentrica: aumento della massa e del volume delle camere; lo spessore (relativo) può essere normale, aumentato o diminuito: cause esercizio isotonico, sovraccarico di volume, perdita di tessuto funzionante per infarto; aggiunta di sarcomeri in serie e allungamento dei miocardiociti. Fra gli atleti, quelli che hanno il cuore più grande sono i rematori, i ciclisti e gli sciatori di fondo; lo spessore di parete in genere non supera 1,3 cm (limite superiore per sedentari 1,1 cm). Gli allenamenti stagionali provocano variazioni stagionali delle dimensioni del cuore.")
5
L’ipertrofia cardiaca da gravidanza regredisce in alcuni mesi dopo il parto.
CURIOSITA’ INTERESSANTE: Nel Pitone di Burma il consumo di un pasto abbondante (vedi Piccolo Principe: il pitone che ha mangiato un elefante) si accompagna ad un aumento del metabolismo di circa 7 volte ed uno straordinario aumento del 40% della massa cardiaca dopo sole 48 ore dal pasto; tutto ritorna alla norma in 28 giorni.
 si accompagna ad un aumento del metabolismo di circa 7 volte ed uno straordinario aumento del 40% della massa cardiaca dopo sole 48 ore dal pasto; tutto ritorna alla norma in 28 giorni.")
6
Al contrario delle modificazioni fisiologiche del cuore, l’ipertrofia patologica è stimolata da attivazione neuroumorale, da un sovraccarico emodinamico cronico o altri fattori di stress per il cuore. Anche il rimodellamento patologico può essere intenso e rapido: la sintesi di MHC (myosin heavy chain) aumenta del 35% poche ore dopo l’esposizione ad un sovraccarico; l’iperespressione di un particolare gene (Akt1) fa aumentare la massa cardiaca del 60% in sole 2 settimane. Nell’ipertrofia cardiaca, le dimensioni dei miocardiociti aumentano e i sarcomeri sono fortemente organizzati (grosse banda al microscopio ottico). L’ipertrofia da esercizio tipicamente non comporta anche un aumento del collagene miocardico. Confrontando l’ipertrofia da esercizio con quella indotta dall’ipertensione i recettori dell’ormone tiroideo e le diverse isoforme della catena pesante della miosina sono regolati in maniera opposta. Le isoforme della miosina sono diverse, e questo può contribuire a ridurre la contrattilità nelle forme patologiche. In ratti sacrificati dopo un allenamento fisico i markers dell’ipertrofia (geni che caratterizzano il fenotipo ipertrofico) sono inibiti.
. L’ipertrofia da esercizio tipicamente non comporta anche un aumento del collagene miocardico. Confrontando l’ipertrofia da esercizio con quella indotta dall’ipertensione i recettori dell’ormone tiroideo e le diverse isoforme della catena pesante della miosina sono regolati in maniera opposta. Le isoforme della miosina sono diverse, e questo può contribuire a ridurre la contrattilità nelle forme patologiche. In ratti sacrificati dopo un allenamento fisico i markers dell’ipertrofia (geni che caratterizzano il fenotipo ipertrofico) sono inibiti.")
7
Gli stimoli di pressione e volume (aumentati) inducono un’espressione diversa di beta-miosina, actina alfa scheletrica e ATPasi del Ca2+ del reticolo sarcoplasmico, anche con simili livelli di ipertrofia. Si è inoltre dimostrato che quello che determina il fenotipo ipertrofico è la natura dello stimolo, non il fatto che sia continuo o intermittente. Visto che l’accrescimento del cuore ad un certo punto si arresta anche se gli stimoli persistono e che regredisce quando gli stimoli vengono rimossi, vi devono essere anche dei meccanismi che si oppongono all’aumento di volume delle cellule. È improbabile che si tratti semplicemente della rimozione degli stimoli. Nel muscolo scheletrico e cardiaco l’attivazione della cascata di segnali che attivano la crescita è accompagnata dall’inibizione di vie che promuovono la proteolisi e il contrario accade nelle condizioni che portano all’atrofia. La plasticità dei miocardiociti è spesso accompagnata dalla reinduzione di un “programma genetico fetale”, nel quale l’espressione genica assomiglia a quella che caratterizza lo sviluppo embrionale.

8
Quando il cuore si scompensa, le pareti ventricolari si assottigliano a causa di una combinazione di proteolisi e morte cellulare. Si era pensato che l’irrorazione sanguigna fosse insufficiente per il miocardio ipertrofico e quindi si sviluppassero condizioni di ischemia, ma questa ipotesi non è stata confermata da tutti gli studi. Si pensa anche all’alterazione delle proteine contrattili, rimodellamento della matrice extracellulare e fibrosi e variazioni dell’attivazione dei recettori beta adrenergici. Più recentemente si è parlato di autofagia, un fatto che riguarda il riciclo delle proteine e degli organelli intracellulari, nella risposta dei miocardiociti allo stress e nella transizione verso l’insufficienza. Diminuzioni della massa cardiaca a livelli ben inferiori al normale si verificano in condizioni di assenza di gravità, nell’allettamento prolungato ed in altre condizioni di scarico ventricolare. In uno studio su soggetti sani allettati per 12 settimane la massa del ventricolo sinistro diminuiva del 15%; in un altro studio, che probabilmente ha raggiunto il limite minimo di atrofia cardiaca, si è documentata una riduzione del 25% della massa in pazienti con lesioni spinali che non potevano muoversi

9
Lo stress meccanico induce segnali paracrini e autocrini regolando la sintesi e la secrezioni di potenti fattori di crescita, fra cui il fattore insulino simile 1 (IGF 1), angiotensina II ed endotelina 1, sia in colture cellulari, sia in pazienti con stenosi aortica. Lo stress meccanico stimola direttamente i recettori per l’angiotensina II, senza che vi sia un aumento della stessa.

10
Risposte cellulari alle alterazioni del carico.
Una complessa sequenza di risposte allo stress neuroumorale e biochimico termina nella regolazione di geni dell’ipertrofia e della crescita cellulare

11
Segnali extracellulari che scatenano risposte intracellulari nel miocardiocita. Questi segnali attivano e sbloccano la trascrizione di geni che presiedono alla crescita cellulare e al rimodellamento. Nonostante la complessità, vi sono dei nodi comuni, sulla superficie cellulare, nel citoplasma e nel nucleo, che potrebbero essere oggetto di interventi terapeutici

12
L’ipertensione è il più importante fattore di rischio di insufficienza cardiaca, a causa del ruolo fondamentale dell’accrescimento miocardico ipertrofico nello sviluppo dell’insufficienza cardiaca. Lo schema prevalente della malattia cardiaca ipertensiva vede prima l’ipertrofia concentrica del ventricolo sinistro, seguita da dilatazione ventricolare e insufficienza contrattile. Vi è una relazione fra l’aumento quantitativo della massa cardiaca e il manifestarsi di situazioni cliniche negative. Oltre alla massa, è importante il tipo di rimodellamento cardiaco: con il rimodellamento concentrico compare una lieve disfunzione sistolica, mentre il malfunzionamento è più rilevante nell’ipertrofia eccentrica.

13
L’ipertrofia dei miocardiociti è la risposta cellulare allo stress biomeccanico, sia esso intrinseco come nell’ipertensione e nei vizi valvolari, o estrinseco come nella cardiomiopatia ipertrofica familiare. L’ipertrofia cardiaca ha l’effetto di normalizzare l’aumento della tensione di parete eliminando lo stimolo iniziale. Gli aspetti caratteristici sono un aumento delle dimensioni dei miocardiociti, un aumento della sintesi proteica e una migliore organizzazione dei sarcomeri. Queste modificazioni del fenotipo cellulare sono precedute e accompagnate dalla reintroduzione del cosiddetto programma genetico fetale. L’ipertrofia in risposta a segnali patologici è di tipo adattativo per mantenere la gettata cardiaca a fronte delle condizioni alterate, ma alla lunga si associa ad un aumento significativo del rischio di morte improvvisa e ad una progressione verso l’insufficienza cardiaca.

14
Il processo ipertrofico non è completamente benefico
Il processo ipertrofico non è completamente benefico. Si solleva il problema se l’ipertrofia da stress miocardico sia un bene oppure se sia di tipo adattativo soltanto in principio ma porti alla perdita funzionale del cuore nel lungo periodo. Altrettanto importante è la differenza fra ipertrofia fisiologica, quella che accompagna lo sviluppo post natale e quella indotta dall’esercizio, e l’ipertrofia patologica. Interventi per favorire la prima ed inibire la seconda potrebbero avere un ovvio valore terapeutico. VIE MOLECOLARI DELL’IPERTROFIA MIOCARDICA Il sistema calcineurina-NFAT. La calcineurina è una fosfatasi serina-treonina, espressa in numerosi tessuti e composta da una subunità catalitica A e una regolatrice B. La calcineurina defosforila fattori di trascrizione della famiglia dei NFAT (nuclear factor of activated T cells), provocando la traslocazione del NFAT nei nuclei e l’attivazione dei geni della risposta immunitaria. Sembra accertato che questa via stia alla base anche dell’ipertrofia cardiaca. Il ruolo della calcineurina nell’ipertrofia dovuta alle cause più comuni sembra accertato. Inoltre, questa via si intreccia con altri importanti segnali che provocano ipertrofia, come quelli controllati dalla glicogeno sintetasi e dal MAPK. Il fatto che la calcineurina sia coinvolta in molti, se non tutti, i processi ipertrofici patologici del cuore ne fa un ovvio bersaglio terapeutico, ma non è chiaro se sia necessario un livello minimo di calcineurina per evitare l’atrofia, dato che essa è implicata anche nell’ipertrofia fisiologica da esercizio. PI3K/Akt/GSK-3-Dependent Signaling. PI3Ks = fosfoinositide 3 chinasi. È una famiglia di enzimi che attivano chinasi proteiche e lipidiche e sono importanti nell’accrescimento, nella sopravvivenza e nella proliferazione delle cellule.
, provocando la traslocazione del NFAT nei nuclei e l’attivazione dei geni della risposta immunitaria. Sembra accertato che questa via stia alla base anche dell’ipertrofia cardiaca. Il ruolo della calcineurina nell’ipertrofia dovuta alle cause più comuni sembra accertato. Inoltre, questa via si intreccia con altri importanti segnali che provocano ipertrofia, come quelli controllati dalla glicogeno sintetasi e dal MAPK. Il fatto che la calcineurina sia coinvolta in molti, se non tutti, i processi ipertrofici patologici del cuore ne fa un ovvio bersaglio terapeutico, ma non è chiaro se sia necessario un livello minimo di calcineurina per evitare l’atrofia, dato che essa è implicata anche nell’ipertrofia fisiologica da esercizio. PI3K/Akt/GSK-3-Dependent Signaling. PI3Ks = fosfoinositide 3 chinasi. È una famiglia di enzimi che attivano chinasi proteiche e lipidiche e sono importanti nell’accrescimento, nella sopravvivenza e nella proliferazione delle cellule.")
16
Il PI3K regola la risposta ipertrofica dei miociti piuttosto che la loro proliferazione. Non ha effetti negativi sulla contrattilità. Numerosi studi basati su ratti transgenici che esprimono in eccesso o in difetto questi enzimi. Uno dei bersagli principali di PI3K è la chinasi serina/treonina Akt, che è attivata attraverso vari stadi di fosforilazione. L’ipertrofia regolata da questi fattori è mediata da GSK-3 e dal “mammalian target of rapamycine (mTor). La rapamicina è un immunosoppressore e quando si lega a mTor blocca la sintesi proteica. Nell’insieme, risulta che GSK-3 integra i segnali di diverse vie ipertrofiche e dev’essere disattivata per la comparsa dell’ipertrofia. Controllo trascrizionale dell’ipertrofia cardiaca mediante MEF2/HDAC. Molte molecole calcio dipendenti, comprese calcineurina, protein chinasi calcio/calmodulina dipendente (CaMK), e MAPKs bastano per produrre ipertrofia e riprogrammare l’espressione genica. Dato che vie diverse evocano simili risposte molecolari, è probabile che le vie ipertrofiche convergano su punti finali comuni. Un candidato probabile è MEF2 (myocyte enhancer factor-2), che rimane a livelli basali di attività trascrizionale nel miocardio adulto ma viene attivato su stimolazione. L’attività MEF2 è controllata dall’associazione diretta con un’istone deacetilasi (HDAC). Stimoli ipertrofici (sovraccarico di pressione) e l’attivazione della calcineurina attivano HDAC, che regola MEF2. Si può concludere che molti, se non tutti, gli stimoli ipertrofici convergono sul nucleo e che le HDAC insieme a MEF2 e forse altri fattori di trascrizione che interagiscono con MEF2, come GATA e NFAT, costituiscono gli integratori di questi segnali.
, e MAPKs bastano per produrre ipertrofia e riprogrammare l’espressione genica. Dato che vie diverse evocano simili risposte molecolari, è probabile che le vie ipertrofiche convergano su punti finali comuni. Un candidato probabile è MEF2 (myocyte enhancer factor-2), che rimane a livelli basali di attività trascrizionale nel miocardio adulto ma viene attivato su stimolazione. L’attività MEF2 è controllata dall’associazione diretta con un’istone deacetilasi (HDAC). Stimoli ipertrofici (sovraccarico di pressione) e l’attivazione della calcineurina attivano HDAC, che regola MEF2. Si può concludere che molti, se non tutti, gli stimoli ipertrofici convergono sul nucleo e che le HDAC insieme a MEF2 e forse altri fattori di trascrizione che interagiscono con MEF2, come GATA e NFAT, costituiscono gli integratori di questi segnali.")
17
Segnali ipertrofici via G Protein–Coupled Receptors (GPCRs) giocano un ruolo importante nella regolazione della funzione cardiaca e negli adattamenti ai carichi emodinamici. I più importanti GPCRs sono i recettori adrenergici e muscarinici. Questi recettori a sette eliche si accoppiano a tre classi principali di proteine eterotrimetriche che legano GTP, che trasducono il segnale agonista o antagonista ad effettori intracellulari (enzimi e canali ionici). Le subunità dei recettori GTP, una volta attivate, si dissociano e attivano segnali intracellulari indipendenti. I recettori di AngII, ET1, alfa-adrenergici si accoppiano all’unità Gq/11 (che a sua volta attiva la fosfolipasi C) e sono tutti sufficienti a provocare ipertrofia. Il recettore adrenergico più abbondante nel cuore è il beta-1, accoppiato a Gs, che attiva l’adenilciclasi, provocando i noti effetti sul cuore (cronotropo, inotropo, lusitropo). La PKA (attivata dalla catena precedente) media le conseguenze negative dell’aumento cronico della stimolazione beta adrenergica, mentre l’adenilciclasi di per se può avere altri effetti protettivi. È stato recentemente dimostrato che il trattamento con beta bloccanti nell’insufficienza cardiaca congestizia è accompagnato anche da un salvataggio del fenotipo dei miocardiociti, con sottoregolazione dei geni dell’ipertrofia e sovraregolazione di geni che erano stati inibiti. Piccole proteine G costituiscono un passaggio critico fra i recettori di membrana e diverse vie di segnalazione. Vi sono molti membri, che regolano diverse funzioni cellulari, fra cui la crescita, la divisione e la sopravvivenza, l’organizzazione del citoscheletro, i traffici transmembrana e la motilità cellulare. Hanno una massa molecolare simile (21 kDa) e si attivano legando il GTP che si idrolizza a GDP e viene disattivato.
 e sono tutti sufficienti a provocare ipertrofia. Il recettore adrenergico più abbondante nel cuore è il beta-1, accoppiato a Gs, che attiva l’adenilciclasi, provocando i noti effetti sul cuore (cronotropo, inotropo, lusitropo). La PKA (attivata dalla catena precedente) media le conseguenze negative dell’aumento cronico della stimolazione beta adrenergica, mentre l’adenilciclasi di per se può avere altri effetti protettivi. È stato recentemente dimostrato che il trattamento con beta bloccanti nell’insufficienza cardiaca congestizia è accompagnato anche da un salvataggio del fenotipo dei miocardiociti, con sottoregolazione dei geni dell’ipertrofia e sovraregolazione di geni che erano stati inibiti. Piccole proteine G costituiscono un passaggio critico fra i recettori di membrana e diverse vie di segnalazione. Vi sono molti membri, che regolano diverse funzioni cellulari, fra cui la crescita, la divisione e la sopravvivenza, l’organizzazione del citoscheletro, i traffici transmembrana e la motilità cellulare. Hanno una massa molecolare simile (21 kDa) e si attivano legando il GTP che si idrolizza a GDP e viene disattivato.")
18
La via MAPK. È un legame importante fra gli stimoli esterni e il nucleo mediante fosforilazione e regolazione di molteplici fattori di trascrizione. Le MAPKs si dividono in 3 sottofamiglie principali: extracellularly responsive kinases (ERKs), c-Jun N-terminal kinases (JNKs), e p38 MAPKs. Gli ultimi due gruppi sono anche chiamati MAPKs che rispondono agli stress perché sono attivati non solo da stimoli anabolici e dagli agonisti delle GPCRs, ma anche da stress patologici come l’ischemia e gli agenti citotossici. PKC e ipertrofia cardiaca: è una chinasi serina/treonina ubiquitaria attivata da recettori accoppiati a proteine G. Varie isoforme sono coinvolte nella patogenesi dell’ipertrofia. L’attività PKC dipende dalla dislocazione spaziale e dall’associazione con proteine del citoscheletro (RACK e AKAPs). Gp130/STAT3 Signaling: è un recettore promiscuo per diverse citochine fra cui interleukin 6/11, leukemia inhibitory factor (LIF), e cardiotrophin-1 (CT-1). CT-1 induce ipertrofia in vitro; ANG II aumenta LIF, CT-1 e IL-6. Metabolismo lipidico e ipertrofia cardiaca. La generazione di combustibile nel cuore adulto si basa sull’ossidazione mitocondriale di acidi grassi a catena lunga per produrre ATP. L’ipertrofia invece si associa alla soppressione dell’ossidazione degli acidi grassi e alla deviazione verso il metabolismo glucidico, che caratterizza il cuore fetale. Questa deviazione si può interpretare come un adattamento perché diminuisce il consumo di ossigeno per mole di ATP generata. Non è però chiaro quali conseguenze metaboliche, come l’accumulo di acidi grassi nel cuore, comporti la soppressione cronica del metabolismo lipidico cardiaco.
. Gp130/STAT3 Signaling: è un recettore promiscuo per diverse citochine fra cui interleukin 6/11, leukemia inhibitory factor (LIF), e cardiotrophin-1 (CT-1). CT-1 induce ipertrofia in vitro; ANG II aumenta LIF, CT-1 e IL-6. Metabolismo lipidico e ipertrofia cardiaca. La generazione di combustibile nel cuore adulto si basa sull’ossidazione mitocondriale di acidi grassi a catena lunga per produrre ATP. L’ipertrofia invece si associa alla soppressione dell’ossidazione degli acidi grassi e alla deviazione verso il metabolismo glucidico, che caratterizza il cuore fetale. Questa deviazione si può interpretare come un adattamento perché diminuisce il consumo di ossigeno per mole di ATP generata. Non è però chiaro quali conseguenze metaboliche, come l’accumulo di acidi grassi nel cuore, comporti la soppressione cronica del metabolismo lipidico cardiaco.")
19
I geni dell’ossidazione degli acidi grassi sono in primo luogo regolati da una famiglia di fattori di trascrizione chiamata peroxisome proliferator-activated receptors (PPARs). I meccanismi della modulazione dipendente da PPAR dell’ipertrofia cardiaca non sono chiari; si indicano due possibilità: a) le alterazioni del metabolismo lipidico nel cuore sono un epifenomeno secondario alle cause della crescita del miocardio; b) le anomalie del metabolismo lipidico precedono e in qualche modo causano l’ipertrofia. La seconda ipotesi è sostenuta dall’osservazione che molte alterazioni ereditarie dell’ossidazione degli acidi grassi si accompagnano ad ipertrofia del ventricolo sinistro. ALTRE VIA DELL’IPERTROFIA CARDIACA Il segnale MMP/TNF. Le MMPs sono enzimi della matrice extracellulare e aumentano nell’ipertrofia post infarto; l’aumento della loro attività contribuisce alla dilatazione progressiva del cuore scompensato, mentre l’inibizione farmacologica migliora l’ipertrofia sperimentale, compresa la funzione contrattile. CHAMP and Cardiomyocyte Hypertrophy. Si tratta di un’elicasi dell’RNA specifica per il cuore, attivata da MEF2. Contribuisce all’ipertrofia. Inibizione dello scambiatore Na/H (NHE). La sua attività aumenta in numerosi modelli di ipertrofia cardiaca e comporta un aumento della concentrazione intracellulare del sodio e, di conseguenza, del calcio perché rallenta lo scambiatorie Na/Ca. L’aumento del calcio intracellulare stimola molte cascate che producono ipertrofia come calcineurin-, CaMK-, PKC- and MAPK-dependent pathways. Quindi l’inibizione di NHE può migliorare molte forme di ipertrofia.
. La sua attività aumenta in numerosi modelli di ipertrofia cardiaca e comporta un aumento della concentrazione intracellulare del sodio e, di conseguenza, del calcio perché rallenta lo scambiatorie Na/Ca. L’aumento del calcio intracellulare stimola molte cascate che producono ipertrofia come calcineurin-, CaMK-, PKC- and MAPK-dependent pathways. Quindi l’inibizione di NHE può migliorare molte forme di ipertrofia.")
20
CARDIAC HYPERTROPHY: COMPENSATORY RESPONSE VERSUS MALADAPTATION
Is Cardiac Hypertrophy Good, Bad, or Ugly? Si ritiene in genere che l’ipertrofia cardiaca possa essere adattativa in alcune situazioni, in particolare negli atleti. È però meno chiaro se la risposta ipertrofica a situazioni patologiche croniche, come l’ipertensione o l’infarto, inizi come risposta compensatoria (che diventa maladattativa solo in seguito) o se questo tipo di accrescimento del miocardio sia dannoso fin dall’inizio. In questo caso le vie molecolari alla base dei due tipi di ipertrofia dovrebbero essere diverse e in effetti ci sono differenze sia morfologiche sia a livello molecolare: l’ipertrofia da esercizio non è accompagnata da accumulo di collagene nel miocardio e in genere l’aumento dello spessore della parete ventricolare sinistra è di grado modesto. Nel confronto fra ratti spontaneamente ipertesi e ratti sottoposti ad esercizio, sono diversi alcuni geni ipertrofici come BNP e ET-1 e l’espressione delle MHCs. In conclusione, l’ipertrofia buona (esercizio) cattiva (patologica) e pessima (scompensata) sono diverse a livello molecolare ma non è escluso che alcune vie ipertrofiche partecipino a tutti i tipi di ipertrofia. INIBIZIONE DELL’IPERTROFIA. Sono stati descritti diversi fattori genetici o esogeni che inibiscono l’ipertrofia: l’inizio e l’inibizione dell’ipertrofia cardiaca coinvolgono segnali multipli formando una specie di rete che si integra e modula una quantità di stimoli. Alcuni modelli di topi transgenici attenuano la risposta ipertrofica al sovraccarico di pressione mantenendo una funzione sistolica normale, mentre modelli diversi sono ugualmente protetti dall’ipertrofia ma hanno una funzione sistolica depressa e un aumento della mortalità. Quindi la risposta ipertrofica può essere dissociata dalla regolazione della contrattilità
 o se questo tipo di accrescimento del miocardio sia dannoso fin dall’inizio. In questo caso le vie molecolari alla base dei due tipi di ipertrofia dovrebbero essere diverse e in effetti ci sono differenze sia morfologiche sia a livello molecolare: l’ipertrofia da esercizio non è accompagnata da accumulo di collagene nel miocardio e in genere l’aumento dello spessore della parete ventricolare sinistra è di grado modesto. Nel confronto fra ratti spontaneamente ipertesi e ratti sottoposti ad esercizio, sono diversi alcuni geni ipertrofici come BNP e ET-1 e l’espressione delle MHCs. In conclusione, l’ipertrofia buona (esercizio) cattiva (patologica) e pessima (scompensata) sono diverse a livello molecolare ma non è escluso che alcune vie ipertrofiche partecipino a tutti i tipi di ipertrofia. INIBIZIONE DELL’IPERTROFIA. Sono stati descritti diversi fattori genetici o esogeni che inibiscono l’ipertrofia: l’inizio e l’inibizione dell’ipertrofia cardiaca coinvolgono segnali multipli formando una specie di rete che si integra e modula una quantità di stimoli. Alcuni modelli di topi transgenici attenuano la risposta ipertrofica al sovraccarico di pressione mantenendo una funzione sistolica normale, mentre modelli diversi sono ugualmente protetti dall’ipertrofia ma hanno una funzione sistolica depressa e un aumento della mortalità. Quindi la risposta ipertrofica può essere dissociata dalla regolazione della contrattilità.")
21
Molti lavori sostengono che l’inibizione dell’ipertrofia cardiaca patologica possa avere effetti benefici anche se lo stimolo originale (aumento dello stress di parete) persiste, ma si tratta di osservazioni a breve termine, mentre è probabile che alla lunga l’inibizione dell’ipertrofia sfoci ugualmente nello scompenso. Qualunque sia l’efficacia di un intervento antiipertrofico, esso deve essere sostenuto nel tempo e non deve compromettere la contrattilità. Bisognerà anche associare strategie complementari, modulando il ciclo del calcio per sostenere la funzione contrattile e inibire le risposte neuroumorali, al fine di trattare con successo e prevenire lo scompenso cardiaco.

22
Antiremodeling Effect of Long-Term Exercise Training in Patients With Stable Chronic Heart Failure
Results of the Exercise in Left Ventricular Dysfunction and Chronic Heart Failure (ELVD-CHF) Trial Pantaleo Giannuzzi, MD; Pier Luigi Temporelli, MD; Ugo Corrà, MD; Luigi Tavazzi, MD; for the ELVD-CHF Study Group (Circulation. 2003;108: ) I volumi telesistolico e telediastolico erano aumentati del 6% e del 7% rispettivamente nel gruppo di controllo, mentre erano diminuiti del 5% e del 9% nel gruppo allenato, nel quale anche la frazione di eiezione era aumentata del 16%. Il tempo di esercizio aumentava nel gruppo allenato di 2,1 min (29%), la capacità di lavoro di 18 W (24%) e il consumo d’ossigeno alla soglia ventilatoria di 2 ml/kg/min (17%). Frequenza cardiaca, pressione sistolica e il prodotto pressione frequenza non cambiavano nei soggetti di controllo ma diminuivano significativamente negli allenati. L’esercizio moderato a lungo termine in pazienti CHF con disfunzione diastolica grave ha un effetto antirimodellamento documentato da una riduzione modesta ma significativa dei volumi del ventricolo sinistro e da un miglioramento della frazione di eiezione. Meccanismi: riduzione della frequenza e della pressione a riposo e a parità di esercizio sottomassimale. Ridotta produzione di ROS nel muscolo scheletrico. Aumento del flusso coronarico nel cuore sano e intorno all’infarto; controllo dell’infiammazione
 Trial. Pantaleo Giannuzzi, MD; Pier Luigi Temporelli, MD; Ugo Corrà, MD; Luigi Tavazzi, MD; for the ELVD-CHF Study Group. (Circulation. 2003;108: ) I volumi telesistolico e telediastolico erano aumentati del 6% e del 7% rispettivamente nel gruppo di controllo, mentre erano diminuiti del 5% e del 9% nel gruppo allenato, nel quale anche la frazione di eiezione era aumentata del 16%. Il tempo di esercizio aumentava nel gruppo allenato di 2,1 min (29%), la capacità di lavoro di 18 W (24%) e il consumo d’ossigeno alla soglia ventilatoria di 2 ml/kg/min (17%). Frequenza cardiaca, pressione sistolica e il prodotto pressione frequenza non cambiavano nei soggetti di controllo ma diminuivano significativamente negli allenati. L’esercizio moderato a lungo termine in pazienti CHF con disfunzione diastolica grave ha un effetto antirimodellamento documentato da una riduzione modesta ma significativa dei volumi del ventricolo sinistro e da un miglioramento della frazione di eiezione. Meccanismi: riduzione della frequenza e della pressione a riposo e a parità di esercizio sottomassimale. Ridotta produzione di ROS nel muscolo scheletrico. Aumento del flusso coronarico nel cuore sano e intorno all’infarto; controllo dell’infiammazione.")
27
Molecular remodeling of cardiac contractile function
JEANNE JAMES AND JEFFREY ROBBINS Am. J. Physiol. 273 (Heart Circ. Physiol. 42): H2105–H2118, 1997 Le proteine dell’apparato contrattile determinano e rispecchiano diverse esigenze fisiologiche per ogni tipo di muscolo. Tutti i muscoli striati hanno un insieme di proteine comuni che sono molto conservate, ma diverse fibre si caratterizzano per particolari e spesso uniche isoforme proteiche complementari. L’esempio più tipico è la catena pesante della miosina (MHC), che appunto esiste in diverse isoforme, caratteristiche di vari fibre muscolari. Esistono isoforme anche della catena leggera della miosina (MLC), dell’actina, del complesso delle troponine, della proteina C che lega la miosina, della titina, della nebulina e della tropomiosina. Nel cuore di mammifero, le diverse isoforme delle proteine contrattili possono essere espresse in risposta a stimoli esterni ed interni e si ritiene che siano responsabili di diversi aspetti funzionali. Per esempio vi sono due isoforme di actina alfa (scheletrica e cardiaca), prodotte da geni simili in cromosomi diversi e presenti entrambe nel cuore. La forma scheletrica è abbondante nel feto ma è sottoregolata nella vita adulta, quando viene sopraregolata l’actina cardiaca. Una forma di topo geneticamente modificato iperesprime l’actina scheletrica nel cuore da adulto e ha un cuore ipercontrattile. La più evidente correlazione fra le proprietà contrattili e le proteine riguarda le MHC: ve ne sono tre isoforme cardiache (V1, V2, V3), in cui cambia la velocità dell’ATPasi e della contrazione. Sono stati studiati diversi tipi di topo transgenico per individuare la correlazione fra caratteristiche funzionali e profilo isomiosinico.
: H2105–H2118, Le proteine dell’apparato contrattile determinano e rispecchiano diverse esigenze fisiologiche per ogni tipo di muscolo. Tutti i muscoli striati hanno un insieme di proteine comuni che sono molto conservate, ma diverse fibre si caratterizzano per particolari e spesso uniche isoforme proteiche complementari. L’esempio più tipico è la catena pesante della miosina (MHC), che appunto esiste in diverse isoforme, caratteristiche di vari fibre muscolari. Esistono isoforme anche della catena leggera della miosina (MLC), dell’actina, del complesso delle troponine, della proteina C che lega la miosina, della titina, della nebulina e della tropomiosina. Nel cuore di mammifero, le diverse isoforme delle proteine contrattili possono essere espresse in risposta a stimoli esterni ed interni e si ritiene che siano responsabili di diversi aspetti funzionali. Per esempio vi sono due isoforme di actina alfa (scheletrica e cardiaca), prodotte da geni simili in cromosomi diversi e presenti entrambe nel cuore. La forma scheletrica è abbondante nel feto ma è sottoregolata nella vita adulta, quando viene sopraregolata l’actina cardiaca. Una forma di topo geneticamente modificato iperesprime l’actina scheletrica nel cuore da adulto e ha un cuore ipercontrattile. La più evidente correlazione fra le proprietà contrattili e le proteine riguarda le MHC: ve ne sono tre isoforme cardiache (V1, V2, V3), in cui cambia la velocità dell’ATPasi e della contrazione. Sono stati studiati diversi tipi di topo transgenico per individuare la correlazione fra caratteristiche funzionali e profilo isomiosinico.")
28
Il resto dell’articolo riguarda le diverse tecniche dell’ingegneria genetica, più che i risultati ottenuti. Si va dall’ablazione di geni specifici alla trasfettazione di porzioni di DNA anche diversi (es Drosofila) e si ottengono topi (tutte le ricerche di questo genere sono state effettuate su topi, per ragioni pratiche anche se sarebbero teoricamente estensibili ad altri animali) che esprimono in quantità aumentata le proteine di cui si vogliono studiare gli effetti. I pezzetti di DNA sono caricati su virus con i quali si infettano le cellule, che poi vengono iniettate su topi genitori, la cui prole sarà portatrice della mutazione. Particolarmente interessante la possibilità di produrre dei topi transgenici nei quali la proteina da studiare viene attivata in un momento voluto, per es tramite tetraciclina e può anche essere disattivata.

30
Per molti anni i cardiologi hanno sostenuto la necessità del riposo prolungato per i pazienti con malattia cardiaca ischemica. Negli ultimi trent’anni, invece, c’è stata una vera rivoluzione e oggi esercizio leggero o moderato è prescritto non solo per la prevenzione ma anche come trattamento fondamentale della malattia. In questa rassegna ci interessiamo del rimodellamento cellulare e molecolare che ha luogo nel cuore in seguito ad allenamento di endurance. Ci riferiamo in particolare alle crescenti dimostrazioni del potenziale rigenerativo del cuore adulto, costituito da una riserva endogena di cellule staminali o progenitrici cardiache.

31
L’accrescimento del cuore (ipertrofia) è in genere definito come fisiologico (normale) o patologico (cattivo). L’ipertrofia cardiaca da esercizio è il prototipo dell’accrescimento fisiologico e si può grossolanamente distinguere in concentrico o eccentrico; la struttura cardiaca è normale e la funzione è normale o migliorata. L’ipertrofia patologica invece si associa alla perdita di miocardiociti (apoptosi e necrosi), sostituzione fibrosa, disfunzione cardiaca e aumento del rischio di insufficienza cardiaca e morte improvvisa. La struttura miocardica è riformata dall’esercizio attraverso un aumento bilanciato della massa miocardica, che comprende l’ipertrofia e la neo angiogenesi (cuore d’atleta). Per esempio, 2 ore al giorno di esercizio su treadmill ad alta intensità (85-90% VO2max) per 5 giorni alla settimana per 8 settimane aumenta del 20-32% e del 17-23% la massa dei miocardiociti in topi maschi e femmine. In genere, gli atleti di endurance (corridori, nuotatori) hanno un aumento di spessore e una notevole dilatazione del ventricolo sinistro (ipertrofia eccentrica), quelli di resistenza (pesisti, lottatori) un aumento notevole dello spessore e una modesta dilatazione (ipertrofia concentrica), mentre quelli “combinati”, che fanno allenamento di resistenza e di endurance (ciclisti, canoisti, rematori) hanno la maggiore ipertrofia. Nonostante i comprovati effetti dell’allenamento nella riduzione del rischio cardiovascolare, in seguito ad episodi di esercizio intenso è stato documentato un aumento della troponina cardiaca, che indica danni miocardici, anche in soggetti sani. Un allenamento di endurance prolungato di alto livello può aumentare il rischio di aritmie e arresto cardiaco.
. Per esempio, 2 ore al giorno di esercizio su treadmill ad alta intensità (85-90% VO2max) per 5 giorni alla settimana per 8 settimane aumenta del 20-32% e del 17-23% la massa dei miocardiociti in topi maschi e femmine. In genere, gli atleti di endurance (corridori, nuotatori) hanno un aumento di spessore e una notevole dilatazione del ventricolo sinistro (ipertrofia eccentrica), quelli di resistenza (pesisti, lottatori) un aumento notevole dello spessore e una modesta dilatazione (ipertrofia concentrica), mentre quelli combinati , che fanno allenamento di resistenza e di endurance (ciclisti, canoisti, rematori) hanno la maggiore ipertrofia. Nonostante i comprovati effetti dell’allenamento nella riduzione del rischio cardiovascolare, in seguito ad episodi di esercizio intenso è stato documentato un aumento della troponina cardiaca, che indica danni miocardici, anche in soggetti sani. Un allenamento di endurance prolungato di alto livello può aumentare il rischio di aritmie e arresto cardiaco.")
32
Per molto tempo si è considerato il cuore un organo post mitotico, privo di capacità rigenerativa, per cui i miocardiociti hanno la stessa età del loro proprietario. Questo non vale per il cuore dei neonati e dei bambini prepuberi: questi cuori giovani hanno una robusta capacità di crescere e di rigenerarsi attraverso la moltiplicazione delle cellule, l’ipertrofia cellulare e dei vasi. Si può raddoppiare la massa ventricolare nel neonato in circa una settimana aumentando il postcarico. Ancora più interessante è il fatto che infarti massivi in soggetti giovani, come quelli che capitano quando una coronaria ha l’origine sbagliata nell’arteria polmonare, dopo correzione chirurgica, rigenerano un miocardio normale con segni minimi o assenti di cicatrice. Negli ultimi dieci anni, fortunatamente, è sorta una nuova era della biologia miocardica e si è fatto spazio il concetto di un cuore di rimpiazzo nell’adulto. Il cuore adulto contiene una riserva di cellule staminali e progenitrici cardiache endogene (eCSC). Queste cellule si riconoscono in base ad uno specifico marcatore (c-kit) e si distinguono da quelle di origine emopoietica (identificate da CD45); hanno le proprietà delle cellule staminali (clonogeniche, auto rinnovanti e multipotenti) sia in vitro sia in vivo. Nell’uomo queste cellule sono attivate e indirizzate verso la linea miocardiocitica in risposta ad aumenti del carico. Utilizzando carbonio radioattivo, un gruppo di ricerca ha dimostrato che almeno metà dei miocardiociti si rigenerano nel corso della vita. Nel ratto, abbiamo riscontrato un aumento della proliferazione, del numero e della differenziazione cardiogenica di eCSC con carichi fisiologici come nuoto o corsa.
. Queste cellule si riconoscono in base ad uno specifico marcatore (c-kit) e si distinguono da quelle di origine emopoietica (identificate da CD45); hanno le proprietà delle cellule staminali (clonogeniche, auto rinnovanti e multipotenti) sia in vitro sia in vivo. Nell’uomo queste cellule sono attivate e indirizzate verso la linea miocardiocitica in risposta ad aumenti del carico. Utilizzando carbonio radioattivo, un gruppo di ricerca ha dimostrato che almeno metà dei miocardiociti si rigenerano nel corso della vita. Nel ratto, abbiamo riscontrato un aumento della proliferazione, del numero e della differenziazione cardiogenica di eCSC con carichi fisiologici come nuoto o corsa.")
33
Cellule staminali cardiache endogene e formazione di nuovi miocardiociti. (A) cellule staminali cardiache isolate da cuore di ratto adulto. (B) un piccolo miocardiocita neoformato (verde, freccia; in rosso actina sarcomerica; nucleo in blu). Aumento del numero di cellule staminali cardaiche in animali allenati al nuoto
 cellule staminali cardiache isolate da cuore di ratto adulto. (B) un piccolo miocardiocita neoformato (verde, freccia; in rosso actina sarcomerica; nucleo in blu). Aumento del numero di cellule staminali cardaiche in animali allenati al nuoto.")
34
Allenamento di endurance provoca anche adattamenti del letto coronarico, con aumento della distribuzione di ossigeno, dell’estrazione, del flusso coronarico, del diametro e del numero delle arterie e miglioramento della funzione endoteliale. I segnali che inducono questi adattamenti sono l’ischemia, fattori di crescita (VEGF e FGF-2) e forze fisiche ed emodinamiche. Inoltre, l’allenamento modifica il numero e le caratteristiche di cellule progenitrici dell’endotelio di origine midollare che contribuiscono alla neo vascolarizzazione. Infine, l’allenamento prolungato migliora la contrattilità dei miocardiociti influendo sul ciclo del calcio. Si ritiene comunemente che stimoli e segnali che conducono all’ipertrofia patologica siano specifici: c’è la riattivazione o sopraregolazione del programma genico di tipo fetale dei cardiomiociti, che comprende aumento di ANP, BNP, actina scheletrica, MLC-1 atriale e b-MHC e diminuzione di geni normalmente espressi nell’adulto, come a-MHC e sarcoplasmic reticulum Ca2+ ATPase (SERCA2a). Tutto questo non avviene in modelli di ipertrofia fisiologica, nella quale agisce una complessa rete di fattori di attivazione, compresi GATA4, GATA6, Csx/Nkx2.5, MEF2, c-jun, c-fos, c-myc, nuclear factor k B and NFAT. La via IGF-1-fosfoinositide 3-chinasi (PI3K) è quella più accreditata per la crescita fisiologica del cuore, mentre recettori accoppiati a proteine G stanno alla base dell’ipertrofia patologica. Con iniezioni intracoronariche di IGF-1 e HGF si è riusciti ad attivare le eCSC e a rigenerare miociti e vasi in una zona infartuata nel maiale, la cui morfologia cardiaca è particolarmente simile a quella dell’uomo. La rigenerazione ha migliorato la sopravvivenza delle cellule, ha ridotto la fibrosi e ha migliorato i parametri funzionali.
. Tutto questo non avviene in modelli di ipertrofia fisiologica, nella quale agisce una complessa rete di fattori di attivazione, compresi GATA4, GATA6, Csx/Nkx2.5, MEF2, c-jun, c-fos, c-myc, nuclear factor k B and NFAT. La via IGF-1-fosfoinositide 3-chinasi (PI3K) è quella più accreditata per la crescita fisiologica del cuore, mentre recettori accoppiati a proteine G stanno alla base dell’ipertrofia patologica. Con iniezioni intracoronariche di IGF-1 e HGF si è riusciti ad attivare le eCSC e a rigenerare miociti e vasi in una zona infartuata nel maiale, la cui morfologia cardiaca è particolarmente simile a quella dell’uomo. La rigenerazione ha migliorato la sopravvivenza delle cellule, ha ridotto la fibrosi e ha migliorato i parametri funzionali.")
36
The name Akt stands for Ak strain transforming
The name Akt stands for Ak strain transforming. The origins of the Akt name date back to 1928, where J. Furth performed experimental studies on mice that developed spontaneous thymic lymphomas. The serine-threonine protein kinase AKT1 is catalytically inactive in serum-starved primary and immortalized fibroblasts. AKT1 and the related AKT2 are activated by platelet-derived growth factor. The activation is rapid and specific, and it is abrogated by mutations in the pleckstrin homology domain of AKT1. It was shown that the activation occurs through phosphatidylinositol 3-kinase. In the developing nervous system AKT is a critical mediator of growth factor-induced neuronal survival. Survival factors can suppress apoptosis in a transcription-independent manner by activating the serine/threonine kinase AKT1, which then phosphorylates and inactivates components of the apoptotic machinery. Mice lacking Akt1 display a 25% reduction in body mass, indicating that Akt1 is critical for transmitting growth promoting signals, most likely via the igf1 receptor. Mice lacking Akt1 are also resistant to cancer:

37
I miocardiociti sono le cellule principali del cuore e anche se sono solo il 20% della popolazione totale di cellule cardiache, costituiscono più del 90% della massa del cuore. Pertanto, la maggior parte delle modificazioni indotte nel cuore dall’allenamento (aerobico) deriva da adattamenti dei miocardiociti e questa plasticità del sistema è alla base di effetti dipendenti dall’intensità (dell’esercizio). I miocardiociti rispondono all’esercizio in molti modi, cioè regolando le proprie dimensioni e la contrattilità e la risposta dipende dall’intensità: maggiore intensità, maggiore adattamento. La crescita adattativa delle cellule in risposta all’esercizio, chiamata ipertrofia fisiologica, si esprime con un aumento proporzionale della sezione e della lunghezza delle cellule, e porta ad un aumento del peso e delle dimensioni dei ventricoli.

38
Con allenamento ad alta intensità (80-90% VO2max) abbiamo ottenuto una risposta ipertrofica proporzionale, già dopo poche settimane di esercizio, che raggiunge un limite dopo due mesi. Sono coinvolti fattori sia trascrizionali sia traslazionali. Queste vie possono avere diversi andamenti temporali e una diversa importanza biologica. L’attivazione della via phosphoinositide-3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) è cruciale per l’induzione dell’ipertrofia fisiologica. Tutto questo aumenta la biogenesi e l’attività dei ribosomi e porta ad un aumento di mRNA e di sintesi proteica. È dimostrato che l’attivazione di questa via fa la differenza fra l’ipertrofia fisiologica e quella patologica.
/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) è cruciale per l’induzione dell’ipertrofia fisiologica. Tutto questo aumenta la biogenesi e l’attività dei ribosomi e porta ad un aumento di mRNA e di sintesi proteica. È dimostrato che l’attivazione di questa via fa la differenza fra l’ipertrofia fisiologica e quella patologica.")
39
Allenamento aerobico di alta intensità (90% VO2max) per periodi prolungati aumenta la contrattilità di miocardiociti scarichi, con un aumento della frazione di accorciamento del 40-50% e della velocità di contrazione/rilasciamento del 20-40%. La potenza aumenta fino al 60%. L’effetto dipende dall’intensità dell’esercizio. Questi miglioramenti sono indipendenti dall’ipertrofia perché la risposta contrattile dipende da meccanismi subcellulari, come l’idrolisi dell’ATP e la formazione di ponti laterali stimolata dal calcio. La curva tensione/lunghezza diventa più ripida. Dato che la gestione del calcio intracellulare controlla la contrazione, non meraviglia che l’allenamento modifichi soprattutto questa funzione. In effetti, il parallelismo fra le più rapidi fasi di salita e di discesa del transiente di calcio e le fasi della contrazione e del rilasciamento suggerisce che vi sia un legame causale. Oltre ad accelerare il ciclo del calcio, l’esercizio diminuisce la concentrazione intracellulare di calcio in diastole, riducendo il rischio di aritmie e la pressione di riempimento. Gli effetti smettono di aumentare dopo circa due mesi di allenamento (o per saturazione o perché bisognerebbe aumentare l’intensità) e si invertono in 2-4 settimane dopo che l’allenamento è terminato. Molti studi dimostrano che l’allenamento aumenta l’mRNA e l’espressione proteica di SERCA2, ma non di fosfolambano (PLB). Quindi aumenta il rapporto SERCA2/PLB lasciando più attiva la pompa sarcoplasmica del calcio. Aumenta anche lo stato di fosforilazione e quindi l’attivazione cronica di CaMKII, che mantiene lo stato di fosforilazione di PLB, che non inibisce più SERCA2. Quindi il calcio sarcoplasmatico viene rimosso più rapidamente e questo spiega la variazione delle cinetiche descritta prima.
 e si invertono in 2-4 settimane dopo che l’allenamento è terminato. Molti studi dimostrano che l’allenamento aumenta l’mRNA e l’espressione proteica di SERCA2, ma non di fosfolambano (PLB). Quindi aumenta il rapporto SERCA2/PLB lasciando più attiva la pompa sarcoplasmica del calcio. Aumenta anche lo stato di fosforilazione e quindi l’attivazione cronica di CaMKII, che mantiene lo stato di fosforilazione di PLB, che non inibisce più SERCA2. Quindi il calcio sarcoplasmatico viene rimosso più rapidamente e questo spiega la variazione delle cinetiche descritta prima.")
40
RICHIAMO DELLA FISIOLOGIA:
SERCA2 - è la pompa che trasporta il calcio dal sarcoplasma al reticolo sarcoplasmatico: responsabile della circolazione di calcio: cisterne terminali > sarcoplasma > (troponina C) > tubuli longitudinali del reticolo SP > cisterne terminali. La circolazione inizia quando vengono attivati i recettori alla rianodina dalla corrente di calcio che attraversa la membrana (canali calcio L) durante il potenziale d’azione e termina quando la cellula si ripolarizza. I SERCA sono particolarmente importanti per la velocità del rilasciamento (effetto lusitropo) (fosfo)lambano (PLB) – è una proteina che blocca i SERCA quando è defosforilata e li libera quando è fosforilata. Per esempio la stimolazione del simpatico aumenta la fosforilazione del PLB
 > tubuli longitudinali del reticolo SP > cisterne terminali. La circolazione inizia quando vengono attivati i recettori alla rianodina dalla corrente di calcio che attraversa la membrana (canali calcio L) durante il potenziale d’azione e termina quando la cellula si ripolarizza. I SERCA sono particolarmente importanti per la velocità del rilasciamento (effetto lusitropo) (fosfo)lambano (PLB) – è una proteina che blocca i SERCA quando è defosforilata e li libera quando è fosforilata. Per esempio la stimolazione del simpatico aumenta la fosforilazione del PLB.")
42
L’esercizio aumenta anche il ciclo intracellulare del calcio, che attiva CaMKII (Ca 2+ /calmodulin-dependent kinase II), che contribuisce a mantenere l’attivazione della sintesi proteica e dell’ipertrofia. Molti passaggi dell’accoppiamento eccitazione-contrazione possono essere influenzati dall’esercizio, sia in condizioni normali che patologiche.

43
Al contrario, non c’è una chiara spiegazione dell’aumento della velocità di salita del calcio sarcoplasmatico durante l’attivazione. È possibile che l’allenamento porti ad un prolungamento cronico del potenziale d’azione e quindi dello stato di eccitamento, almeno in alcune parti del cuore oppure che aumenti la sensibilità dei recettori alla rianodina alla corrente di calcio attraverso i canali L, attivati dal potenziale d’azione. In ogni caso, più che la quantità di calcio durante l’attivazione, cambia la forma della curva della concentrazione, che diventa più stretta. Si suppone che questo indirizzi più efficacemente il calcio verso la troponina, aumentando la sincronizzazione dell’attivazione dei sarcomeri. E questa è una spiegazione più credibile dell’aumento di contrattilità. Vi è anche un miglior controllo del pH intracellulare e una trasformazione verso isoforme delle proteine contrattili più efficienti

44
Il modello animale più usato per studiare l’ipertrofia patologica è il ratto post infarto. Viene chiusa permanentemente una coronaria e a questo segue un infarto di dimensioni ripetibili, con conseguente perdita della funzione di pompa, ipertrofia e dilatazione. I miocardiociti del ratto infartuato hanno le caratteristiche opposte a quelle dell’ipertrofia da esercizio. Minore velocità di accorciamento e rilasciamento, minore forza contrattile, alterata gestione del calcio intracellulare e aumento del calcio libero in diastole. Tutte queste alterazioni sono suscettibili di miglioramento in seguito all’allenamento. Innanzitutto, è invertita la disfunzione arteriosa grazie al ripristino della produzione di NO da parte dell’endotelio, una modificazione facilitata da alterazioni adattative dell’eNOS, attivato da Akt e da una riduzione delle specie reattive dell’ossigeno, generate dalla NADPH ossidasi. Questo avviene nelle arterie, ma riduce il carico sul cuore e ne migliora la funzione. Ma ancora più importante è l’attenuazione della disfunzione cardiaca intrinseca. Nell’insieme, questi effetti riducono l’ipertrofia patologica. Giocano un effetto positivo tutti i meccanismi visti per l’ipertrofia fisiologica, dall’aumento della contrattilità e della fase di rilasciamento alla migliore gestione del calcio intracellulare. Infine, l’esercizio corregge i diversi difetti metabolici del cuore dilatato

45
Sono stati escogitati diversi modelli animali per simulare disfunzioni miocardiche non dipendenti dal post infarto. In particolare, un tipo di diabete e la sindrome metabolica. L’allenamento modula positivamente le funzioni contrattili intrinseche, riconducendole a livelli quasi normali. Il principale meccanismo è una normalizzazione del ciclo del calcio.

46
Tutti gli studi descritti indicano che la capacità di rispondere all’allenamento è mantenuta anche durante lo sviluppo di miopatie cardiache e insufficienza dovute all’infarto, al diabete o alla sindrome metabolica e che i meccanismi coinvolti restano simili a quelli del cuore normale. È importante notare che l’esercizio corregge i difetti di inotropismo e lusitropismo direttamente nei miocardiociti e che questo non dipende da fattori esterni alle cellule. In realtà, le alterazioni geniche indotte da diversi tipi di malattia che portano all’ipertrofia patologica sono molteplici e diverse da quelle che stanno alla base della risposta fisiologica. Probabilmente è proprio per questo che tale risposta è conservata anche in condizioni patologiche. IMPLICAZIONI CLINICHE Recentemente è stato dimostrato che programmi d’esercizio ad alta intensità (90% VO2 max) possono essere adeguati per pazienti con ipertrofia post infarto, malattia coronarica o aumentato rischio cardiovascolare. Tutti questi programmi si basavano su una frequenza cardiaca pari al 95% del massimo teorico e hanno ottenuto effetti molto superiori a quelli di esercizi moderati, che aumentano la capacità d’esercizio ma fanno poco sul cuore.
 possono essere adeguati per pazienti con ipertrofia post infarto, malattia coronarica o aumentato rischio cardiovascolare. Tutti questi programmi si basavano su una frequenza cardiaca pari al 95% del massimo teorico e hanno ottenuto effetti molto superiori a quelli di esercizi moderati, che aumentano la capacità d’esercizio ma fanno poco sul cuore.")
47
Cardiac remodeling and failure From molecules to man (Part II)
Paul W.M. Fedak T , Subodh Verma, Richard D. Weisel, Ren-Ke Li Division of Cardiac Surgery, University of Toronto, Toronto General Hospital, 14EN-215, 200 Elizabeth Street, Toronto ON, Canada M5G 2C4 Cardiovascular Pathology 14 (2005) 49 – 60 La matrice extracellulare (ECM) della maggior parte dei tessuti consiste di una complessa rete di collagene fibrillare, elastina, proteine miofibrillari, proteoglicani e protene di adesione, come la laminina e la fibronectina. L’ECM costituisce l’impalcatura fisica dell’organizzazione tridimensionale delle cellule e determina le proprietà fisiche dei tessuti: è un tessuto dinamico che modifica composizione e disposizione in risposta a stimoli ambientali o a lesioni. I tessuti metabolicamente attivi, compresi miocardio e vasi, ricambiano in continuazione il loro ECM. L’ECM non si limita a fornire un supporto strutturale alle cellule, ma è un microambiente dinamico che trasmette segnali alle cellule dallo spazio interstiziale e pertanto contribuisce alla regolazione delle funzioni cellulari. Pertanto, l’interazione delle cellule con la matrice circostante condiziona in vario modo il rimodellamento cardiovascolare, regolando anche la differenziazione, la proliferazione, la crescita e la sopravvivenza dei miocardiociti. Se gli elementi della matrice sono insufficienti, l’abbattimento delle interazioni ECM-cellule porta alla loro morte per apoptosi. Queste osservazioni significano che la composizione, l’orientamento e l’abbondanza degli elementi della matrice del cuore possono avere un’influenza preponderante sulla struttura e la funzione del miocardio.
 49 – 60. La matrice extracellulare (ECM) della maggior parte dei tessuti consiste di una complessa rete di collagene fibrillare, elastina, proteine miofibrillari, proteoglicani e protene di adesione, come la laminina e la fibronectina. L’ECM costituisce l’impalcatura fisica dell’organizzazione tridimensionale delle cellule e determina le proprietà fisiche dei tessuti: è un tessuto dinamico che modifica composizione e disposizione in risposta a stimoli ambientali o a lesioni. I tessuti metabolicamente attivi, compresi miocardio e vasi, ricambiano in continuazione il loro ECM. L’ECM non si limita a fornire un supporto strutturale alle cellule, ma è un microambiente dinamico che trasmette segnali alle cellule dallo spazio interstiziale e pertanto contribuisce alla regolazione delle funzioni cellulari. Pertanto, l’interazione delle cellule con la matrice circostante condiziona in vario modo il rimodellamento cardiovascolare, regolando anche la differenziazione, la proliferazione, la crescita e la sopravvivenza dei miocardiociti. Se gli elementi della matrice sono insufficienti, l’abbattimento delle interazioni ECM-cellule porta alla loro morte per apoptosi. Queste osservazioni significano che la composizione, l’orientamento e l’abbondanza degli elementi della matrice del cuore possono avere un’influenza preponderante sulla struttura e la funzione del miocardio.")
48
I fibroblasti sono le cellule principali del miocardio e hanno lo scopo di sintetizzare e regolare la composizione dell’ECM. L’elevato numero di fibroblasti nel cuore indica che l’ECM è regolato dinamicamente e suggerisce che esso debba avere importanti effetti sulla funzione cardiaca. Il collagene fibrillare nel cuore è organizzato in strati che circondano gruppi di miocardiociti e li collegano fra loro. Lo strato più esterno, l’epimisio circonda tutti i gruppi di miocardiociti del cuore. L’onnipresente perimisio descrive lo strato di tessuto connettivo che circonda e collega gruppi di miocardiociti in una particolare direzione; si può studiare in microscopia ottica o confocale. L’endomisio è la parte di matrice più interna che avvolge le singole cellule cardiache di un gruppo; si vede solo al microscopio elettronico. L’epimiso e il perimisio sono costituiti soprattutto da connettivo fibrillare, mentre l’endomisio è un complesso agglomerato di collagene ed elastina. È chiaro che il contenuto totale e la disposizione di questi elementi della matrice, soprattutto la rete di collagene fibrillare, controllano la forma e la distensibilità dell’intero miocardio. L’orientamento spaziale del perimisio si modifica a seconda delle condizioni di carico del cuore, assumendo una struttura a spirale quando è rilasciato e lineare quando è stirato passivamente. Le fibre del perimisio si orientano secondo l’asse lungo dei gruppi di miociti che avvolgono, ma forniscono anche estese ramificazioni e interconnessioni che legano insieme i miocardiociti in tutto il miocardio, formando una rete organizzata di fibrille connettive.

49
È evidente che le capacità contrattili del miocardio rappresentano un importante esempio di ingegneria biologica, realizzato dalla sua struttura tridimensionale che è largamente basata sul contenuto e l’organizzazione della rete di fibrille collagene. Rimodellamento del collagene in CHF. Molte malattie cardiovascolari, compreso l’infarto miocardico, l’ipertensione e l’insufficienza cardiaca, si associano ad alterazioni della quantità, del tipo, della stabilità e dell’organizzazione del collagene fibrillare. Oltre alle modificazioni cellulari, anche la ristrutturazione della matrice extracellulare gioca un ruolo centrale nel rimodellamento del cuore insufficiente e nella transizione finale verso lo scompenso. La deposizione di nuovo collagene (aumento della quantità totale) è una risposta classica al danno tessutale e serve come supporto strutturale e rinforzo delle zone danneggiate e indebolite. Sotto questo punto di vista, all’interno di una zona di intensa necrosi cellulare, l’aumento del collagene rappresenta un meccanismo adattativo. Il collagene non aumenta con un sovraccarico puro di volume. Se la quantità di collagene è certamente importante, altrettanto lo sono la sua composizione e qualità. Il cuore umano contiene soprattutto collagene di tipi I e III: il primo ha la resistenza alla tensione dell’acciaio e costituisce l’85% del totale. La sua presenza aumenta lo spessore del perimisio e questo è in relazione con la capacità del collagene fibrillare di sostenere lo stiramento e di coordinare la contrazione globale dell’organo. Il collagene di tipo III è meno rigido e conferisce elasticità al miocardio.
 è una risposta classica al danno tessutale e serve come supporto strutturale e rinforzo delle zone danneggiate e indebolite. Sotto questo punto di vista, all’interno di una zona di intensa necrosi cellulare, l’aumento del collagene rappresenta un meccanismo adattativo. Il collagene non aumenta con un sovraccarico puro di volume. Se la quantità di collagene è certamente importante, altrettanto lo sono la sua composizione e qualità. Il cuore umano contiene soprattutto collagene di tipi I e III: il primo ha la resistenza alla tensione dell’acciaio e costituisce l’85% del totale. La sua presenza aumenta lo spessore del perimisio e questo è in relazione con la capacità del collagene fibrillare di sostenere lo stiramento e di coordinare la contrazione globale dell’organo. Il collagene di tipo III è meno rigido e conferisce elasticità al miocardio.")
50
In tutti i tessuti e organi, compreso il cuore, le fibre collagene si stabilizzano maturando, con la formazione di legami crociati biochimici: il grado di interconnessione del collagene è probabilmente più importante della quantità e del rapporto fra le diverse isoforme, per il rimodellamento cardiaco. Nel miocardio normale la maglia di collagene fibrillare è molto confluente, ampia e distribuita omogeneamente. Ogni fibra del perimisio è abbondante e relativamente spessa. È dimostrato che nel cuore insufficiente la maglia collagene è alterata e degradata e le fibre del perimisio sono di meno e più sottili. Turnover del collagene L’ECM del cuore ha una notevole plasticità: si stima che gli elementi delle diverse componenti del collagene siano degradati e rimpiazzati al ritmo dello 0,6% al giorno, con una semivita delle proteine compresa fra 80 e 120 giorni. Quindi, le componenti dell’ECM sono continuamente sintetizzate e degradate, anche se lentamente nel cuore normale Le MMPs sono un’importante famiglia di enzimi endogeni dipendenti dallo zinco che degradano l’ECM di quasi tutti i tessuti biologici: sono coinvolte nel rimodellamento fisiologico dei tessuti nello sviluppo, la morfogenesi, la riproduzione e l’infiammazione, così come nel rimodellamento patologico. Se ne conoscono più di 20 specie, che degradano la maggior parte delle proteine dell’ECM, ma ognuna ha una speciale affinità per singole componenti della matrice ed agiscono in maniera organizzata e coordinata durante il processo di rimodellamento.

51
Le MMPs sono classificate in 4 gruppi a seconda della loro specificità primaria per le componenti della matrice, cioè: le collagenasi (MMP-1 e MMP-13), le gelatinasi (MMP-2 eMMP-9), le stromolisine (MMP-3), e le MMPs di membrana (MT1-MMP). La maggior parte delle MMPs sono costitutive e la loro espressione è altamente inducibile in risposta a diversi fattori quali lo stiramento meccanico, i fattori di crescita, le citochine e altri peptidi bioattivi che si trovano nel miocardio, in particolare nel processo dell’insufficienza cadiaca. L’espressione delle MMPs è iniziata in risposta a segnali ambientali e in effetti la maggior parte di esse si trovano nell’ECM in uno stato latente di proenzimi (pro-MMP) e sono attivate rapidamente. Pertanto, le MMPs rispondono a stimoli ambientali in maniera rapida e sostenuta in modo da riorganizzare la struttura e il contenuto dell’ECM nel tessuto che le contiene. Si ritiene che nel miocardio sia i fibroblasti sia le mast cellule sintetizzino e secernano quasi tutte le MMPs nello spazio extracellulare. L’espressione e l’attività delle MMPs aumentano nell’insufficienza cardiaca di diversa eziologia, perché il rimodellamento cardiaco da parte di questi enzimi costituisce una risposta deliberata e comune al danno tessutale. Regolazione dell’attività delle MMPs Svariati processi regolano l’equilibrio dell’attività delle MMPs: l’espressione e la secrezione delle MMPs nell’ECM, l’attivazione di MMPs latenti e l’inibizione competitiva da parte di inibitori endogeni. Il risultato netto di tutti questi processi determina l’attività enzimatica complessiva nel compartimento extracellulare e il destino dell’ECM.
 e sono attivate rapidamente. Pertanto, le MMPs rispondono a stimoli ambientali in maniera rapida e sostenuta in modo da riorganizzare la struttura e il contenuto dell’ECM nel tessuto che le contiene. Si ritiene che nel miocardio sia i fibroblasti sia le mast cellule sintetizzino e secernano quasi tutte le MMPs nello spazio extracellulare. L’espressione e l’attività delle MMPs aumentano nell’insufficienza cardiaca di diversa eziologia, perché il rimodellamento cardiaco da parte di questi enzimi costituisce una risposta deliberata e comune al danno tessutale. Regolazione dell’attività delle MMPs. Svariati processi regolano l’equilibrio dell’attività delle MMPs: l’espressione e la secrezione delle MMPs nell’ECM, l’attivazione di MMPs latenti e l’inibizione competitiva da parte di inibitori endogeni. Il risultato netto di tutti questi processi determina l’attività enzimatica complessiva nel compartimento extracellulare e il destino dell’ECM.")
52
A livello dell’espressione genica delle MMPs e della secrezione nell’ECM, si sa che le citochine e altri fattori di crescita aumentano nell’insufficienza cardiaca, e sono importanti induttori genetici. In particolare le citochine proinfiammatorie, come il TNF-, sono coinvolte sia nell’espressione delle MMPs, sia nella riduzione dei loro inibitori specifici, e questo provoca un aumento generalizzato delle MMPs. TIMPs: presentazione e concetti chiave Un interessante sviluppo delle conoscenze sulle MMPs è stata la scoperta di una famiglia di 4 inibitori endogeni delle MMPs, denominati TIMPs, che rappresentano il sistema endogeno primario che controbilancia dinamicamente l’attività delle MMPs e sono probabilmente il punto di regolazione primario nei mammiferi. Il miocardio umano è in qualche modo unico, nel senso che esprime, quando è sano, tutte e quattro le specie di TIMPs. Tutte insieme, le diverse TIMPs si legano saldamente a quasi tutte le MMPs e le inattivano. Si considerano ora le TIMPs proteine multifunzionali dotate di diversi effetti biologici e varie modalità d’azione. Per esempio, le proteine TIMP sono coinvolte nella crescita, proliferazione e apoptosi delle cellule, e talvolta i loro effetti non dipendono dalle MMPs. Il profilo dell’espressione delle TIMPs è alterato nel cuore insufficiente: è stata descritta una riduzione della metà dell’espressione della TIMP-1 in associazione con il degrado della matrice e con la disfunzione miocardica in seguito a danno da ischemia-riperfusione. Il controllo del rimodellamento dell’ECM dovrebbe fornirci l’anello mancante nell’attuale insufficiente armamentario terapeutico nei CHF e una migliore comprensione del ruolo delle proteine TIMP nel miocardio normale e insufficiente dovrebbe indirizzare la messa a punto di specifiche strategie anti rimodellamento.

53
Cardiac remodelling: concentric versus eccentric hypertrophy in strength and endurance athletes
C. Mihl, W.R.M. Dassen, H. Kuipers Netherlands Heart Journal, Volume 16, Number 4, April 2008 Ci siamo chiesti in che modo l’ipertrofia cardiaca indotta dall’esercizio differisca da quella di origine patologica. • il rimodellamento fisiologico è un’alterazione compensatoria sia delle proporzioni sia della funzione del cuore: si verifica negli atleti; • il rimodellamento patologico accompagna stati come l’infarto miocardico (sovraccarico da pressione), infiammazioni miocardiche, la miocardiopatia dilatativa o il sovraccarico da volume. La distensione provocata dall’aumento del carico emodinamico inizia la risposta ipertrofica del cuore. I miocardiociti si espandono sintetizzando nuove proteine contrattili e assemblando nuovi sarcomeri in parallelo, aumentando la forza contrattile della cellula. Questo tipo di rimodellamento è omogeneo. Quando il cuore è sottoposto ad un aumento cronico delle richieste funzionali (come dopo un infarto) ha luogo un diverso tipo di rimodellamento, che è per lo più irreversibile. Questo tipo di rimodellamento, associato a sovraccarico cronico, si associa ad un contributo sproporzionato e inomogeneo dei fibroblasti che aumentano il collagene fibrillare nell’interstizio
, infiammazioni miocardiche, la miocardiopatia dilatativa o il sovraccarico da volume. La distensione provocata dall’aumento del carico emodinamico inizia la risposta ipertrofica del cuore. I miocardiociti si espandono sintetizzando nuove proteine contrattili e assemblando nuovi sarcomeri in parallelo, aumentando la forza contrattile della cellula. Questo tipo di rimodellamento è omogeneo. Quando il cuore è sottoposto ad un aumento cronico delle richieste funzionali (come dopo un infarto) ha luogo un diverso tipo di rimodellamento, che è per lo più irreversibile. Questo tipo di rimodellamento, associato a sovraccarico cronico, si associa ad un contributo sproporzionato e inomogeneo dei fibroblasti che aumentano il collagene fibrillare nell’interstizio.")
54
Questo provoca la perdita di miocardiociti per apoptosi o necrosi e le cellule morte sono sostituite da fibroblasti e collagene extracellulare. La fibrosi aumenta la rigidità del cuore interferendo con il riempimento diastolico. La perdita di miocardiociti è un fattore importante per l’insufficienza cardiaca: l’apoptosi riduce la forza contrattile e assottiglia le pareti, portando alla dilatazione miocardica. Si distinguono due diverse forme del cuore d’atleta: un cuore allenato alla forza e uno allenato alla resistenza. Aumenta il diametro del ventricolo sinistro e lo spessore di parete

55
Nell’allenamento aerobico, il fattore dominante è il sovraccarico di volume e quindi si sviluppa un’ipertrofia eccentrica. L’aumento della pressione arteriosa nel sollevamento di pesi può arrivare a 320/250 mmHg. Livelli così elevati di postcarico e pressione ventricolare aumentano lo sforzo (stress) di parete e questo è il principale stimolo per l’ipertrofia nel cuore sovraccaricato. Il cuore risponde all’aumento dello sforzo di parete nel training di forza aggiungendo nuovi sarcomeri in parallelo a quelli già esistenti. Aumenta così lo spessore della parete e si ha l’ipertrofia concentrica. Negli sport di endurance, come ciclismo, maratona e triatlon, prevale il rimodellamento eccentrico, soprattutto nei ciclisti. Gli effetti del lavoro isometrico dipendono da due fattori, cioè l’intensità dell’esercizio e l’entità delle masse muscolari coinvolte. Negli atleti di resistenza aumenta lo spessore di parete e anche un poco il volume ventricolare, ma lo stress di parete rimane invariato. Una meta analisi della funzione e della struttura cardiaca di ciclisti, corridori e atleti di forza nonché soggetti di controllo non ha trovato differenze per quanto riguarda la massa ventricolare degli atleti (ma nei controlli era minore); il diametro ventricolare era diverso nei tre gruppi rispetto ai controlli, mentre lo spessore relativo di parete era minore negli atleti di endurance che in quelli di forza (0.39 mm vs mm).
 di parete e questo è il principale stimolo per l’ipertrofia nel cuore sovraccaricato. Il cuore risponde all’aumento dello sforzo di parete nel training di forza aggiungendo nuovi sarcomeri in parallelo a quelli già esistenti. Aumenta così lo spessore della parete e si ha l’ipertrofia concentrica. Negli sport di endurance, come ciclismo, maratona e triatlon, prevale il rimodellamento eccentrico, soprattutto nei ciclisti. Gli effetti del lavoro isometrico dipendono da due fattori, cioè l’intensità dell’esercizio e l’entità delle masse muscolari coinvolte. Negli atleti di resistenza aumenta lo spessore di parete e anche un poco il volume ventricolare, ma lo stress di parete rimane invariato. Una meta analisi della funzione e della struttura cardiaca di ciclisti, corridori e atleti di forza nonché soggetti di controllo non ha trovato differenze per quanto riguarda la massa ventricolare degli atleti (ma nei controlli era minore); il diametro ventricolare era diverso nei tre gruppi rispetto ai controlli, mentre lo spessore relativo di parete era minore negli atleti di endurance che in quelli di forza (0.39 mm vs mm).")
56
CONCLUSIONI: lo sviluppo di un cuore d’atleta di endurance oppure di resistenza non va considerato obbligatorio. Atleti di forza, che avrebbero dovuto sviluppare un’ipertrofia concentrica pura, avevano il diametro ventricolare aumentato; atleti di resistenza, per i quali ci si aspettava un’ipertrofia eccentrica pura avevano un pronunciato aumento dello spessore di parete.

57
Physiological Society Symposium – The Athlete’s Heart
Exercise-induced cardiac hypertrophy: a substrate for sudden death in athletes? G. Hart Experimental Physiology (2003) 88.5, 639–644 Negli studi sperimentali, l’indice più comunemente usato per stabilire la presenza e la gravità dell’ipertrofia è il rapporto fra il peso del cuore e quello corporeo. Indagini ecocardiografihe nell’uomo hanno dimostrato che l’aumento dello spessore della parete del ventricolo sinistro è normalmente distribuito in maniera concentrica attorno alla cavità della camera, benché si possa riscontrare un’ipertrofia asimmetrica negli atleti fino al 12% dei casi, con un aumento sproporzionato del setto. Finora si considera che l’aumento del volume del cuore sia dovuto essenzialmente ad ipertrofia, piuttosto che ad iperplasia. Ogni aumento delle dimensioni dei miocardiociti implica un’alterazione delle caratteristiche elettriche delle cellule, a causa della maggiore superficie del doppio strato dielettrico del sarcolemma.
 88.5, 639–644. Negli studi sperimentali, l’indice più comunemente usato per stabilire la presenza e la gravità dell’ipertrofia è il rapporto fra il peso del cuore e quello corporeo. Indagini ecocardiografihe nell’uomo hanno dimostrato che l’aumento dello spessore della parete del ventricolo sinistro è normalmente distribuito in maniera concentrica attorno alla cavità della camera, benché si possa riscontrare un’ipertrofia asimmetrica negli atleti fino al 12% dei casi, con un aumento sproporzionato del setto. Finora si considera che l’aumento del volume del cuore sia dovuto essenzialmente ad ipertrofia, piuttosto che ad iperplasia. Ogni aumento delle dimensioni dei miocardiociti implica un’alterazione delle caratteristiche elettriche delle cellule, a causa della maggiore superficie del doppio strato dielettrico del sarcolemma.")
58
Alterazioni funzionali del miocardiocita
In un’ipertrofia di media entità è stato descritto un aumento modesto, oppure nullo, dell’ampiezza della contrazione, mentre nell’ipertrofia importante, accompagnata o meno da insufficienza cardiaca, l’ampiezza della contrazione è normalmente ridotta e la durata è aumentata. Sono state descritte modificazioni a tutti i livelli nel processo di accoppiamento eccitazione-contrazione nell’ipertrofia. A livello del sarcolemma, la risposta più consistente è un prolungamento della durata del potenziale d’azione (APD), che, all’opposto di quel che riguarda le proprietà meccaniche, è comune a tutti i tipi di ipertrofia. Il prolungamento dell’APD dipende da un’alterazione dell’equilibrio delle correnti di membrana nella fase di plateau, con un aumento delle correnti in ingresso o una riduzione di quelle in uscita, o più frequentemente con entrambi. La variazione più frequente nell’ipertrofia è una riduzione della corrente transitoria verso l’esterno di potassio (Ito). L’APD dei miociti ventricolari cambia nello spessore del ventricolo: è più lunga nello strato sub-endocardico: nell’ipertrofia, la durata del potenziale d’azione non cambia, o addirittura diminuisce, nei miociti sub-endocardici, mentre aumenta negli strati intermedi e sub-epicardici. Comunque, per la maggior parte dei miociti che compongono la massa ventricolare, l’aumento dell’APD rimane la risposta principale all’ipertrofia.
, che, all’opposto di quel che riguarda le proprietà meccaniche, è comune a tutti i tipi di ipertrofia. Il prolungamento dell’APD dipende da un’alterazione dell’equilibrio delle correnti di membrana nella fase di plateau, con un aumento delle correnti in ingresso o una riduzione di quelle in uscita, o più frequentemente con entrambi. La variazione più frequente nell’ipertrofia è una riduzione della corrente transitoria verso l’esterno di potassio (Ito). L’APD dei miociti ventricolari cambia nello spessore del ventricolo: è più lunga nello strato sub-endocardico: nell’ipertrofia, la durata del potenziale d’azione non cambia, o addirittura diminuisce, nei miociti sub-endocardici, mentre aumenta negli strati intermedi e sub-epicardici. Comunque, per la maggior parte dei miociti che compongono la massa ventricolare, l’aumento dell’APD rimane la risposta principale all’ipertrofia.")
59
Meccanismi cellulari alla base delle aritmie nell’ipertrofia cardiaca
Il prolungamento dell’intervallo QT, sia esso dovuto ad una delle sindromi congenite di QT lungo, o alla somministrazione di particolari farmaci che bloccano le correnti ripolarizzanti, predispone ad un tipo di tachicardia ventricolare detto «torsade de pointes» e alla morte improvvisa. Oggi si sa che il prolungamento dell’APD e il blocco delle correnti ripolarizzanti nell’ipertrofia cardiaca possono predisporre ad aritmie e morte improvvisa, analogamente alla sindrome del QT lungo. Elettrofisiologia cellulare in seguito ad allenamento Un esercizio di resistenza regolare induce molte modificazioni adattative nel sistema cardiovascolare, come l’espansione del volume plasmatico, la bradicardia a riposo, l’aumento del tono vagale, la riduzione del tono simpatico, l’inibizione dei riflessi barocettivi, l’aumento della gittata sistolica e l’aumento della circolazione coronarica. Molte delle risposte adattative positive del cuore degli atleti, che aumentano la contrattilità e la potenza, hanno luogo a livello del miocardiocita, anche se non mancano importanti contributi del sistema nervoso autonomo e della circolazione periferica. L’allenamento di resistenza porta ad un aumento della lunghezza e della capacitanza dei miocardiociti; tuttavia le variazioni morfometriche dei miocardiociti su un modello di ratto sottoposto ad esercizio volontario non sono risultate uniformi: le dimensioni delle cellule epicardiche non cambiavano, mentre quelle sub-endocardiche aumentavano di ampiezza e di volume, suggerendo che le prime modificazioni elettriche dovute all’allenamento abbiano luogo in questo strato.

60
TORSADE DE POINTES

61
Alterazioni elettrocardiografiche negli atleti
Le variazioni elettrocardiografiche che si riscontrano nei pazienti con ipertrofia cardiaca da ogni causa consistono in un aumento di ampiezza delle deflessioni ventricolari (che rappresentano il cosiddetto criterio del voltaggio per diagnosticare l’ipertrofia ventricolare), alterazioni dell’onda T, con inversione del segmento ST, e battiti extrasistolici ventricolari premature più frequenti e complessi. L’intervallo QT, che corrisponde alla durata della depolarizzazione ventricolare, è normale o appena aumentato nel cuore ipertrofico. Questo richiede una spiegazione, dato che a livello delle cellule il prolungamento dell’APD è sempre marcato e ben documentato in tutte le forme di ipertrofia: probabilmente dipende dalla distribuzione alterata della ripolarizzazione. Nel cuore normale, l’APD è più lungo negli strati endocardici, mentre invece nell’ipertrofia l’APD dura di più negli starti intermedi e superficiali e quindi i miocardiociti che danno origine alla parte terminale dell’onda T sono quelli endocardici nel cuore normale, ma cedono il passo a quelli più superficiale nell’ipertrofia. In conseguenza di tutto ciò, l’ipertrofia cardiaca può prolungare poco l’intervallo QT elettrocardiografico, anche se rimane una «sindrome del QT lungo» a livello della maggior parte, ma non di tutti, i miocardiociti.
, alterazioni dell’onda T, con inversione del segmento ST, e battiti extrasistolici ventricolari premature più frequenti e complessi. L’intervallo QT, che corrisponde alla durata della depolarizzazione ventricolare, è normale o appena aumentato nel cuore ipertrofico. Questo richiede una spiegazione, dato che a livello delle cellule il prolungamento dell’APD è sempre marcato e ben documentato in tutte le forme di ipertrofia: probabilmente dipende dalla distribuzione alterata della ripolarizzazione. Nel cuore normale, l’APD è più lungo negli strati endocardici, mentre invece nell’ipertrofia l’APD dura di più negli starti intermedi e superficiali e quindi i miocardiociti che danno origine alla parte terminale dell’onda T sono quelli endocardici nel cuore normale, ma cedono il passo a quelli più superficiale nell’ipertrofia. In conseguenza di tutto ciò, l’ipertrofia cardiaca può prolungare poco l’intervallo QT elettrocardiografico, anche se rimane una «sindrome del QT lungo» a livello della maggior parte, ma non di tutti, i miocardiociti.")
62
Importanza clinica dell’ipertrofia cardiaca nell’allenamento
C’è una certa sovrapposizione fra l’ipertrofia dell’atleta e quella dovuta ad ipertensione, sia rispetto all’entità, sia alla sua reversibilità e alle variazione della geometria ventricolare. Un’ipertrofia moderata da cause patologiche, come quella da esercizio, può associarsi ad una funzione contrattile aumentata oppure normale, ma sostanzialmente le caratteristiche elettriche del tessuto e delle cellule dell’ipertrofia da esercizio sono simili a quelle dell’ipertrofia da altre cause. Nell’ipertrofia da esercizio non siamo in grado di distinguere variazioni legate ad un aumento delle funzioni normali da quelle associate a condizioni patologiche che comportano la morte improvvisa. È pertanto opportuno considerare l’ipertrofia da esercizio nello spettro delle risposte cellulari al sovraccarico cardiaco da qualunque causa e valutare le possibili conseguenze fisiopatologiche in questo quadro. I dati della ricerca di Framingham e di altri studi hanno dimostrato chiaramente che la mortalità per cause cardiache è aumentata dall’ipertrofia di qualunque entità. La morte improvvisa negli atleti è rara e i dati pubblicati indicano che la maggior parte degli eventi è legata a malattie cardiache pre-esistenti, gravi ma non diagnosticate e asintomatiche, che possono essere congenite o acquisite, per esempio patologia coronarica, alterazioni specifiche del muscolo cardiaco (cardiomiopatia) e infiammazione del cuore (miocardite). Rimane però il fatto che nel 15% dei casi, soprattutto nei più giovani, l’unica anomalia riscontrabile è l’ipertrofia. Le aritmie ventricolari frequenti e complesse che sono comuni negli atleti allenati si spiegano sulla base di specifiche alterazioni soltanto in un terzo dei casi.
 e infiammazione del cuore (miocardite). Rimane però il fatto che nel 15% dei casi, soprattutto nei più giovani, l’unica anomalia riscontrabile è l’ipertrofia. Le aritmie ventricolari frequenti e complesse che sono comuni negli atleti allenati si spiegano sulla base di specifiche alterazioni soltanto in un terzo dei casi.")
63
In conclusione, per la maggior parte degli atleti allenati le conseguenze cardiache della loro attività hanno caratteristiche adattative, benigne e probabilmente reversibili. Rimane il fatto che allenamenti atletici intensivi sono associati con un piccolo ma evidente aumento del rischio di morte improvvisa, che può dipendere dalle alterazioni elettriche a livello cellulare che accompagnano l’ipertrofia di grado medio o moderato.

64
The athlete’s heart David Oakley Sheffield, UK Heart 2001;86:722–726 Sull’argomento del cuore d’atleta, bisogna distinguere fra atleti d’élite, maschi e femmine che fanno attività ricreazionale, non atleti che desiderano mantenere una buona efficacia cardiovascolare e pazienti atleti con malattie cardiache diagnosticate. Un programma di allenamento regolare porta a cambiamenti favorevoli nella performance del muscolo scheletrico e a due chiari effetti cardiovascolari – cioè l’ingrandimento del cuore e una bassa frequenza a riposo: queste sono le componenti di un quadro clinico caratteristico, definito «cuore d’atleta». Gli atleti nell’insieme consultano frequentemente il medico lamentando palpitazioni, capogiro, affaticamento, dolore toracico e dispnea esagerata. La visita può mettere in luce alcuni segni inconsueti, che non tranquillizzeranno il medico, ben consapevole dei casi clamorosi di morte improvvisa durante le attività sportive, e il reperto di un ECG apparentemente alterato aumenterà la preoccupazione. Se le dimensioni cardiache sono corrette per la superficie corporea e o per la massa magra, diventa più difficile dimostrare l’ipertrofia concentrica negli atleti di resistenza. Ci sono molti dati sull’entità di queste variazioni e su cosa deve essere considerato compreso nei valori normali per un atleta. Anche se le conclusioni sono svariate, un’utile meta analisi di Fagart sui dati disponibili indica che uno spessore di parete superiore a 1.6 cm è raro per un atleta sano e che per lo più è meno di 1,3 cm.

65
I canoisti, i ciclisti e gli sciatori di fondo sono quelli che hanno il cuore più grande, ma il grado di ipertrofia non correla con l’intensità dell’esercizio o con la performance degli atleti. Molti campioni olimpici hanno dimensioni cardiache normali, quando studenti che fanno sport possono avere un’ipertrofia pronunciata. Questa osservazione significa che la risposta cardiaca all’allenamento non è dovuta solo allo sforzo emodinamico durante l’esercizio, mentre entrano in gioco altri fattori, come stimoli ormonali e predisposizione genetica. È infatti sempre più chiaro che la tendenza all’ipertrofia è in parte determinata geneticamente e che l’esercizio è un fattore scatenante. Studi su gemelli identici confermano questa ipotesi. Qualunque sia il meccanismo, è evidente che l’ipertrofia è una risposta diretta all’allenamento. Atleti che si allenano periodicamente hanno variazioni stagionali del volume ventricolare. Dopo disallenamento si assiste spesso alla regressione dell’ipertrofia, anche dopo molti anni di allenamento. La dilatazione cardiaca e l’ipertrofia possono essere tali da assomigliare a stati patologici, ma gli indici della funzione ventricolare, sia sistolica sia diastolica, sono sostanzialmente normali. Non ci sono prove che soggetti sani possano allenarsi al punto da indursi condizioni patologiche, come fibrosi e disallineamento delle fibre. La bradicardia a riposo è caratteristica dell’atleta allenato: in casi eccezionali po’ scendere sotto a 40 battiti al minuto. Se cuori denervati battono più lentamente dopo allenamento, la bradicardia degli atleti è dovuta soprattutto ad aumento del tono vagale e ad una riduzione di quello simpatico a riposo.

66
La bradicardia a riposo può predisporre ad un aumento di attività ectopiche atriali o ventricolari e in alcuni casi alla fibrillazione atriale. L’aumentare dell’età può modificare alcuni degli adattamenti di cui abbiamo parlato. Molti atleti si tengono allenati fino alla mezza età e partecipano a competizioni «master» e «veterani». Ci sono prove che l’allenamento continuato migliora le funzioni diastoliche negli anziani, ma in uno studio sono stati visti problemi al ventricolo sinistro in ciclisti giapponesi veterani, anche se non fu possibile stabilire se fossero dovuti all’allenamento o all’insorgere di altre patologie, per esempio alle coronarie. È stato anche visto che la bradicardia è più pronunciata negli anziani, in cui è anche ridotta la frequenza massima; tutto questo può contribuire alla riduzione della performance. Gli atleti possono riferire sintomi, che in genere hanno una spiegazione benigna – giramenti di testa da disidratazione o da ipotensione post-esercizio, dolore toracico da tracheite o dolore muscolo scheletrico (in particolare negli sport di contatto e in allenamenti molto pesanti), palpitazioni da contrazioni premature benigne e mancanza di fiato per infezioni polmonari o broncospasmo da esercizio. Ciononostante è spesso necessaria un’indagine approfondita per garantire al paziente e al suo medico la sicurezza di cui hanno bisogno.
, palpitazioni da contrazioni premature benigne e mancanza di fiato per infezioni polmonari o broncospasmo da esercizio. Ciononostante è spesso necessaria un’indagine approfondita per garantire al paziente e al suo medico la sicurezza di cui hanno bisogno.")
67
L’ovvia preoccupazione del medico consultato da un atleta è «sto trascurando un problema cardiaco che potrebbe essere mortale?». In un giovane atleta con ipertrofia o bradicardia eccessivi, la diagnosi differenziale si pone con la cardiomiopatia dilatativa o con la sindrome da seno patologico (sick sinus). Non c’è test che possa garantire la differenza fra cuore d’atleta e miocardiopatia dilatativa, anche se possono aiutare l’entità e la simmetria dell’ipertrofia all’ecocardiogramma, la risposta a tre mesi di riposo, il reperto di ipertrofia non spiegata nei familiari, la risposta pressoria e metabolica all’esercizio e altre caratteristiche ecocardiografiche, come i profili di riempimento del ventricolo sinistro. La morte di un giovane atleta ben preparato è estremamente rara, con un’incidenza di 1: , ma diverse indagini danno esiti molto variabili. Le morti sono più frequenti in giocatori dilettanti e di mezza età, ma anche in questi gruppi il rischio è molto basso, e comunque minore che nella popolazione sedentaria. I casi di morte sono però più frequentemente associati all’esercizio che a periodi di riposo. La morte può essere non cardiaca – per esempio traumatica, da ipo o iper termia, da disidratazione o da abuso di droghe. Rimane il fatto che la morte improvvisa è comunque soprattutto cardiovascolare e un’autopsia ben fatta in genere rivela una causa sottostante. Nei giovani, la cardiomiopatia dilatativa è la causa più frequente, ma alcuni sostengono che la displasia aritmogena del ventricolo destro sia ancora più comune. Fra altre cause vi sono l’origine anomala delle coronarie, la stenosi aortica, la miocardite, la sindrome di Marfan, la sindrome di Wolff-Parkinson-White, la sindrome del QT lungo, il prolasso della mitrale e malattie coronariche.

68
Non vi è uno sport che abbia il monopolo delle morti improvvise, anche se lo squash è stato indicato come particolarmente a rischio, forse a causa della sua natura molto vigorosa e competitiva. Sta crescendo l’interesse per gli sport estremi (per esempio triatlon, corse di 24 ore, fell running): lo stato di esaurimento fisico completo di alcuni partecipanti comporta alterazioni metaboliche che influenzano negativamente la performance cardiaca. Il fatto che all’autopsia di atleti morti improvvisamente si riscontrino frequentemente alterazioni patologiche indica che avrebbe potuto essere compiuta una diagnosi precedente delle loro condizioni nel corso di screening di routine, e che un trattamento appropriato e giusti consigli avrebbero potuto evitare la morte. In alcune nazioni, come l’Italia, lo screening è obbligatorio ed è dimostrato che il programma è efficace. C’è però chi critica questa pratica sostenendo che si cercano malformazioni rare, per le quali mancano trattamenti risolutivi e gli esami non sono perfetti. Anche se si istituisce un programma di screening, non ci sono linee guida riconosciute su come debba essere organizzato e con quali esami. Gli esami che abbiamo a disposizione non sono abbastanza sensibili e specifici, mentre la probabilità che ci siano alterazioni cardiache è molto bassa: questo comporta inevitabilmente diagnosi mancate e, ancor peggio, numerosi falsi positivi. La diagnosi errata di un problema cardiaco in un atleta normale è disastrosa per una persona per la quale lo stato di salute è, per definizione, della massima importanza. Una volta sollevato il dubbio di un problema cardiaco, è molto difficile superarlo. Mentre la disputa sui vantaggi dello screening continua, è evidente che ogni sintomo o segno sospetto in un atleta richiede le indagini più approfondite.

Presentazioni simili

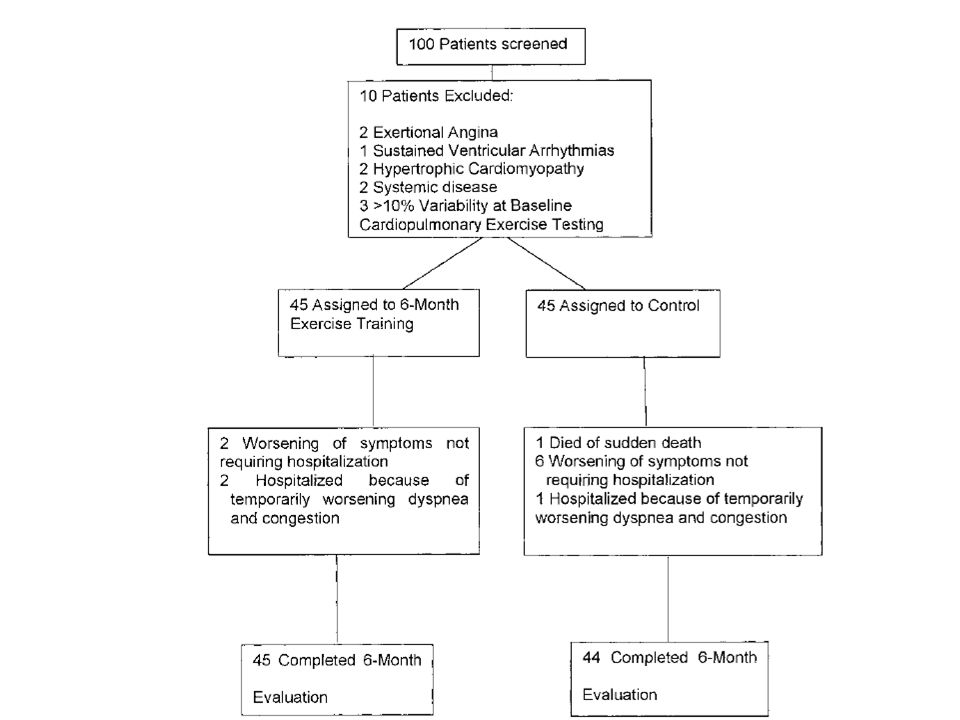
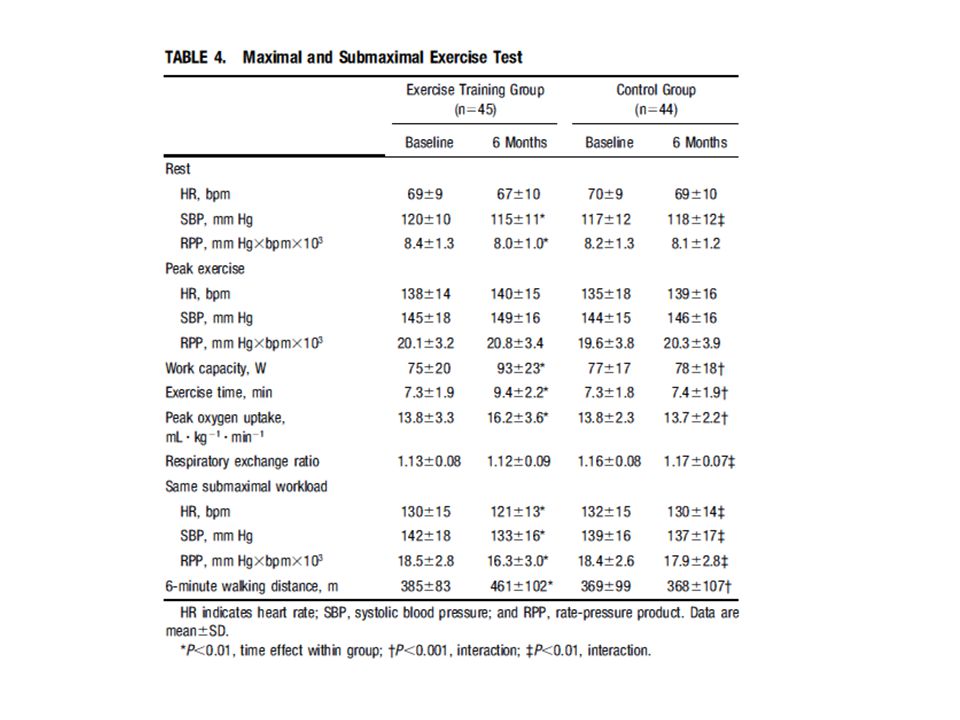
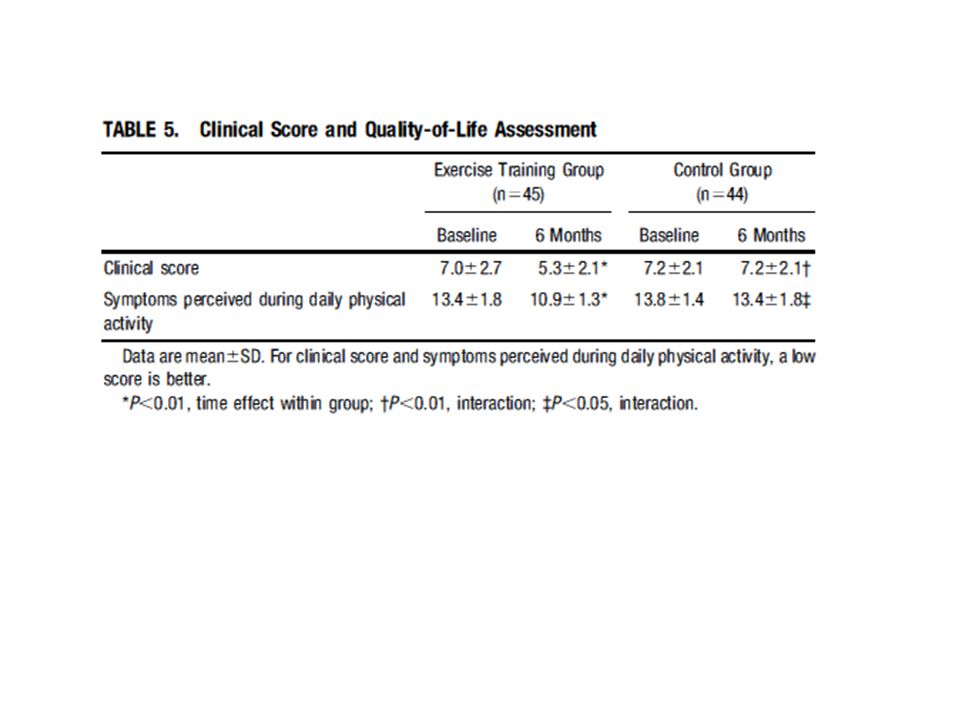
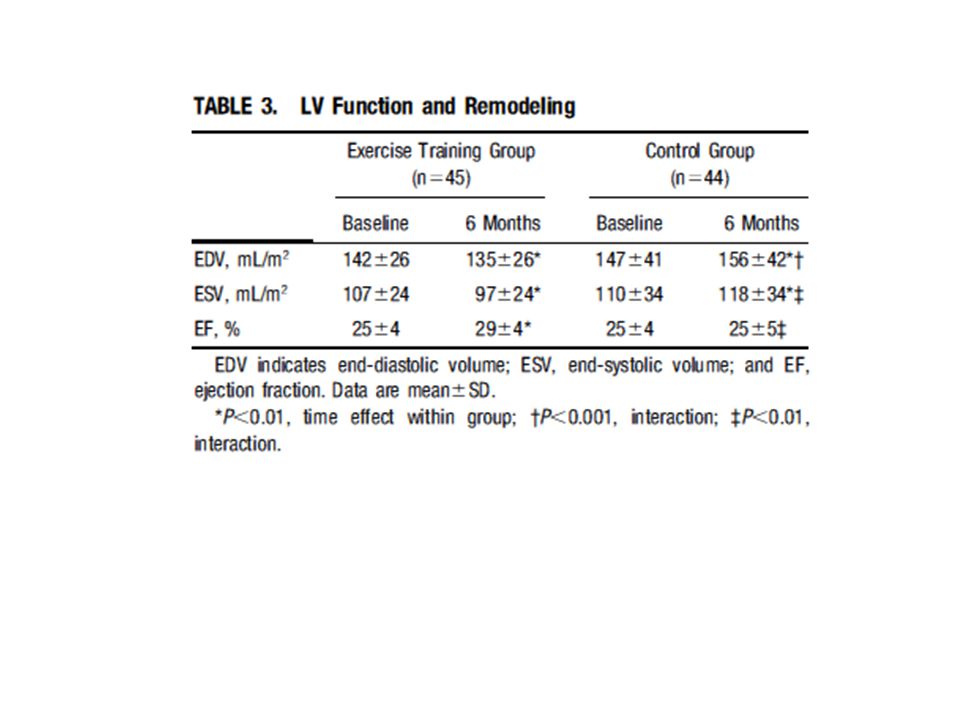

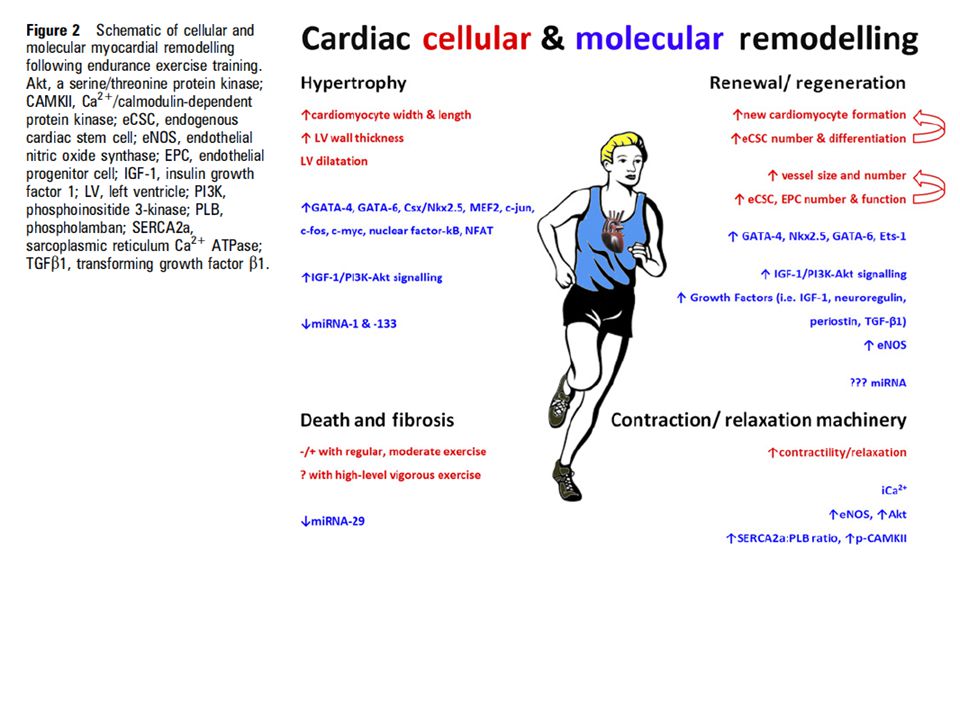
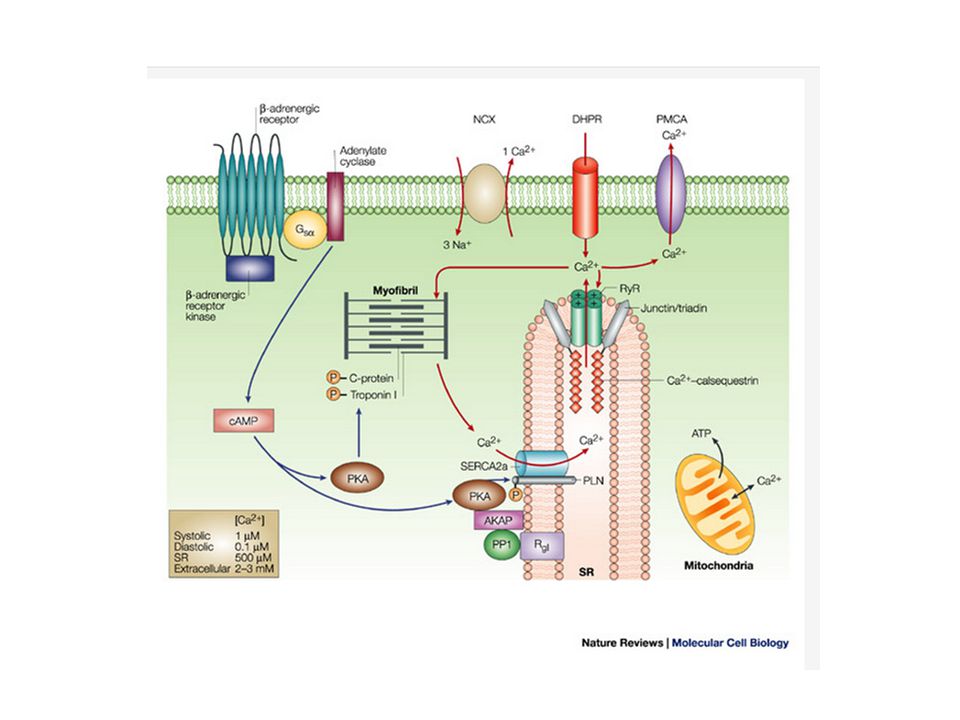


 (Ito) (IKr, IKs) (ICa-L) (INa).>")











>")




