Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Allineamento multiplo
Before about 1800, attempts at trying to uncover the secrets held by the ancient Egyptian hieroglyphics found on walls inside numerous tombs was useless. The pictures were falsely believed to be symbolic, representing some sort of object or idea. Something soon changed all that. The year 1799 was the beginning of an amazing breakthrough for Egyptology. This was the year in which French troops stumbled across the Rosetta Stone near a town in Lower Egypt known as Rosetta. The black basalt stone was inscribed with three different forms of writing: Egyptian hieroglyphics, demotic (a short-hand of hieroglyphics), and Greek written in 196 BC. The Rosetta Stone then became a key element in the decipherment of an Egyptian form of writing known as hieroglyphics. Biochimica computazionale
, and Greek written in 196 BC. The Rosetta Stone then became a key element in the decipherment of an Egyptian form of writing known as hieroglyphics. Biochimica computazionale.")
2
Allineamenti multipli
Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente più rilevanti tendono a mantenersi invariati nelle proteine omologhe, mentre i siti meno importanti possono cambiare anche molto. Osservare e studiare le conservazioni significa capire come le famiglie di proteine funzionano, cosa la rende diverse tra loro, se esistono o meno relazioni filogenetiche inter e intrafamiglia. In questo modo è possibile individuare la funzione di una proteina ignota solo osservando la sequenza dei suoi residui.
, ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente più rilevanti tendono a mantenersi invariati nelle proteine omologhe, mentre i siti meno importanti possono cambiare anche molto. Osservare e studiare le conservazioni significa capire come le famiglie di proteine funzionano, cosa la rende diverse tra loro, se esistono o meno relazioni filogenetiche inter e intrafamiglia. In questo modo è possibile individuare la funzione di una proteina ignota solo osservando la sequenza dei suoi residui.")
3
Applicazioni dell’allineamento multiplo
‘Assemblaggio’ dei genomi Primers per PCR Consensi, motivi Profili, modelli markoviani Definizione di famiglie Filogenesi Caonsensi e motivi sono rappresentazioni sintetiche della conservazione Profili e modelli markoviani sono rappresentazioni probabilistiche Inferenze strutturali Inferenze funzionali Biochimica computazionale

4
Similitudine e omologia
Omologia: carattere QUALITATIVO che posseggono quelle sequenze che derivano da un antenato comune in seguito al processo evolutivo. O due geni sono omologhi o non lo sono. Non esiste una percentuale di omologia. Similitudine: carattere QUANTITATIVO che origina da un allineamento. Il grado di identità che si determina tra i residui allineati o il fatto che residui simili possano corrispondere in un allineamento, può essere quantificato disponendo di metri di valutazione oggettivi, come le matrici di sostituzione. => un’alta similitudine tra proteine può essere indice di omologia, ma non si può escludere il contrario. Esistono infatti proteine molto simili in organismi filogeneticamente non correlati tra loro e proteine molto diverse che possono essere ricondotte a omologhe mediante altri studi

5
Geni ortologhi e geni paraloghi
Geni ortologhi: geni simili riscontrabili in organismi correlati tra loro. Il fenomeno della speciazione porta alla divergenza dei geni e quindi delle proteine che essi codificano. es. l’ α-globina di uomo e di topo hanno iniziato a divergere circa 80 milioni di anni fa, quando avvenne la divisione che dette vita ai primati e ai roditori. I due geni sono da considerarsi ortologhi. Geni paraloghi: geni originati dalla duplicazione di un unico gene nello stesso organismo. es. α-globina e β-globina umana hanno iniziato a divergere in seguito alla duplicazione di un gene globinico ancestrale. I due geni sono da considerarsi paraloghi.

6
Le sequenze da multiallineare in genere si ottengono dalla ricerca in banca dati mediante i sistemi di ricerca per similarità come BLAST e FASTA. Visto che derivano già da un allineamento (anche se prodotto con metodi euristici) e visto che si prendono in considerazione solo sequenze che hanno un alto score (o un basso E, expectation value), l’allineamento mutiplo su questi DATASET darà risultati soddisfacenti. In un allineamento multiplo si prendono in considerazione le colonne di residui, più che le proteine a cui appartengono. Ogni residuo incolonnato è da considerarsi in modo implicito come evolutivamente correlato, in qualche modo.
 e visto che si prendono in considerazione solo sequenze che hanno un alto score (o un basso E, expectation value), l’allineamento mutiplo su questi DATASET darà risultati soddisfacenti. In un allineamento multiplo si prendono in considerazione le colonne di residui, più che le proteine a cui appartengono. Ogni residuo incolonnato è da considerarsi in modo implicito come evolutivamente correlato, in qualche modo.")
7
Significato biologico dell’allineamento multiplo
L’allineamento multiplo riassume La storia evolutiva di una famiglia di proteine La conservazione dei residui dipendente dalla funzione La conservazione dei residui dipendente dalla struttura

8
Allineamenti multipli Vs. allineamenti a coppie
G F P V N I T H S D K L Q R Y M - W C 1: 2: 3: 4: 5: 6: 1: 2: 3: 4: 5: 6: B

9
Significato funzionale della conservazione
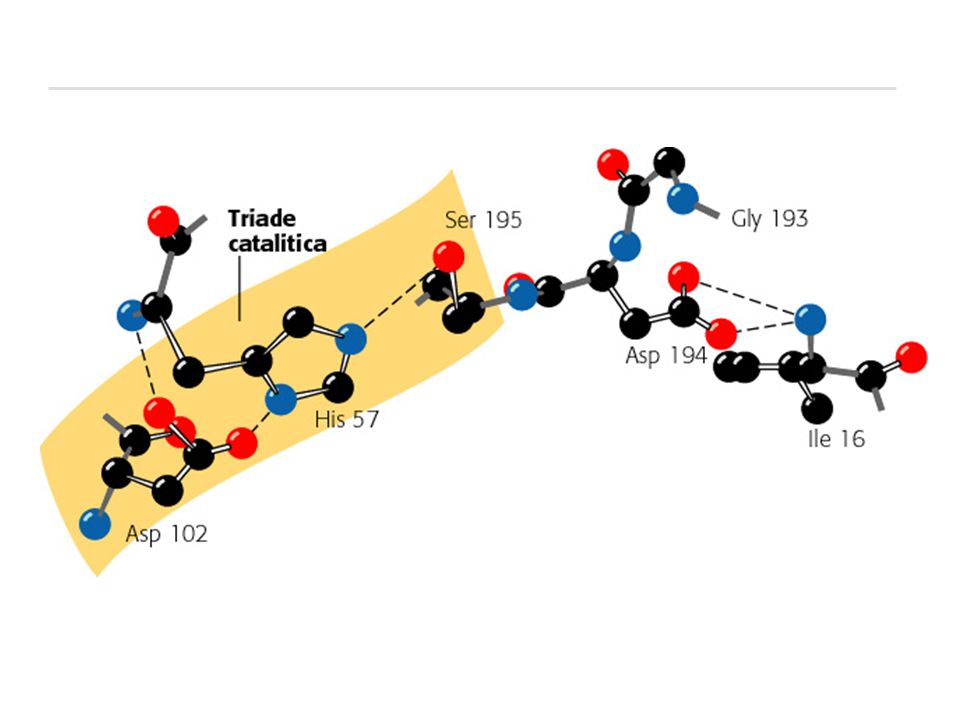
Y I _ D O M E : N K P G / 8 H B V L 1 U S C A Q W - F 4 9 5 6 7 3 2 Conservazione degli amino acidi catalitici in alcuni membri della famiglia della tripsina

11
Significato strutturale dell’allineamento multiplo
PDBSUM of 1tlk a b c C d e f g Sequenze molto divergenti del domino immunoglobulinico allineate manualmente sulla base dell’informazione della struttura di uno dei membri della famiglia (PDB 1tlk). Si noti la presenza di inserzioni o delezioni soprattutto in regioni corrispondenti a loop. Due residui di cisteina che formano un ponte disolfuro nel core idrofobico della proteina sono conservati in tutti i componenti della famiglia.
. Si noti la presenza di inserzioni o delezioni soprattutto in regioni corrispondenti a loop. Due residui di cisteina che formano un ponte disolfuro nel core idrofobico della proteina sono conservati in tutti i componenti della famiglia.")
12
Somiglianze locali di struttura
Fungal/bacterial Phospholipase A2 Eli et al. EMBO, 2001 Ca2+ loop Active site Phospholipase A2 active site Una regione conservata delle fosfolipasi A2 da funghi e batteri è allineabile con altre sequenze della famiglia. Inferenze strutturali e funzionali possono essere fatte sulla base della struttura della sequenza di cobra (PDB 1a3d)
")
13
Allineamenti strutturalmente “corretti”
Divergenza strutturale (RMS) Divergenza di sequenza Relazione tra la divergenza in struttura e in sequenza. La conservazione della struttura è sempre maggiore della conservazione di sequenza. Somiglianze che sono difficilmente riscontrabili in sequenza possono emergere chiaramente da un confronto strutturale. Un allineamento strutturalmente “corretto” non è sempre possibile. Solo il 50% dei residui è allineabile strutturalmente in proteine che hanno tra il 20% e il 30% di identità
 Divergenza di sequenza. Relazione tra la divergenza in struttura e in sequenza. La conservazione della struttura è sempre maggiore della conservazione di sequenza. Somiglianze che sono difficilmente riscontrabili in sequenza possono emergere chiaramente da un confronto strutturale. Un allineamento strutturalmente corretto non è sempre possibile. Solo il 50% dei residui è allineabile strutturalmente in proteine che hanno tra il 20% e il 30% di identità.")
14
Significato evolutivo dell’allineamento multiplo
In linea di principio esiste sempre un allineamento evolutivamente “corretto”. Tuttavia, non esiste un riferimento indipendente (come nel caso delle strutture cristallografiche), e la storia evolutiva delle sequenze deve anzi essere inferita dall’allineamento
, e la storia evolutiva delle sequenze deve anzi essere inferita dall’allineamento.")
15
Difficoltà dell’allineamento multiplo
Complessità del problema Tempo di elaborazione Funzione oggettiva di punteggio ”Peso” da assegnare alle varie sequenze

16
Complessità dell’allineamento multiplo
Sulla superficie del cubo si hanno le matrici di confronto a coppie tra le sequenze A-B, B-C e A-C. L’allineamento ottimale di tre sequenze (A-B-C) richiede il riempimento del cubo e la valutazione di tutte le possibili mosse all’interno del cubo. La complessità di questo algoritmo è (O(LN)), dove L è la lunghezza e N il numero delle sequenze. Per tre sequenze di 300 amino acidi il numero di confronti è 2.7 x 107. Un algoritmo completo di programmazione dinamica è utilizzabile solo nel caso di tre sequenze.
 richiede il riempimento del cubo e la valutazione di tutte le possibili mosse all’interno del cubo. La complessità di questo algoritmo è (O(LN)), dove L è la lunghezza e N il numero delle sequenze. Per tre sequenze di 300 amino acidi il numero di confronti è 2.7 x 107. Un algoritmo completo di programmazione dinamica è utilizzabile solo nel caso di tre sequenze.")
17
Funzione oggettiva per l’allineamento multiplo
Seq1 A A A A Seq2 A A A A Seq3 A A A C Seq4 A A C C Seq1 Seq3 Seq Seq2 Seq Seq4 AAAA AAAC AAAA AAAC AAAA AAAC AACC AAAA AACC AAAA Seq2 Seq4 Somma delle coppie N(N-1)/2 confronti Possibile relazione filogenetica delle sequenze Viene usato il metodo della somma delle coppie in ogni colonna per determinare il punteggio totale dell’allineamento. Questo metodo non tiene in considerazione la storia delle sequenze e il fatto che uno stesso carattere nella colonna può essere facilmente condiviso da sequenze molto simili per ragioni evolutive. Per ovviare a questo si assegna un peso alle sequenze in modo da aumentare il punteggio in confronti tra sequenze evolutivamente distanti e diminuirlo in confronti tra sequenze vicine.
/2 confronti. Possibile relazione filogenetica delle sequenze. Viene usato il metodo della somma delle coppie in ogni colonna per determinare il punteggio totale dell’allineamento. Questo metodo non tiene in considerazione la storia delle sequenze e il fatto che uno stesso carattere nella colonna può essere facilmente condiviso da sequenze molto simili per ragioni evolutive. Per ovviare a questo si assegna un peso alle sequenze in modo da aumentare il punteggio in confronti tra sequenze evolutivamente distanti e diminuirlo in confronti tra sequenze vicine.")
18
Metodi per l’allineamento multiplo
Allineamento ottimale Programmazione dinamica, MSA Allineamento euristico Progressivo globale (CLUSTALW,Pileup) locale (PIMA) Iterativo globale (PRRP) locale (DIALIGN)
 locale (PIMA) Iterativo. globale (PRRP) locale (DIALIGN)")
19
Allineamento con programmazione dinamica
Carrillo & Lipmann, 1988 Per trovare un allineamento ottimale tra tre sequenze è necessario solo calcolare i punteggi all’interno del volume in grigio il volume è delimitato dalle proiezioni delle aree sulle facce del cubo. Le aree sulle facce del cubo sono definite da due segmenti. Uno è il percorso dall’allineamento ottimale a coppie, l’altro la proiezione dell’allineamento multiplo euristico. Questo algoritmo (implementato nel programma MSA) può essere utilizzato per poche (<10) sequenze corte.
 può essere utilizzato per poche (<10) sequenze corte.")
20
N sequenze (dataset) disposte a caso, non allineate
Determinare un albero guida basato sui punteggi di similarità di tutte le coppie Allineare tutte le proteine con tutte le proteine, a coppie ( N(N-1)/2 allineamenti) A partire dalla coppia più simile, determinare le colonne conservate, e allineare la coppia successiva mantenendo queste colonne e ricalcolando lo score complessivo N sequenze (dataset) allineate
/2 allineamenti) A partire dalla coppia più simile, determinare le colonne conservate, e allineare la coppia successiva mantenendo queste colonne e ricalcolando lo score complessivo. N sequenze (dataset) allineate.")
21
Allineamento progressivo
Allineamento multiplo Allineamento progressivo DGEKFGPPQRSGQRSG |||.||| ||||.||| DGERFGP-QRSGNRSG SeqA SeqB Allineamento della coppia A-B Allineamento di tre sequenze A, B, C per passi successivi consenso DHEKFGSSQRSGQRSG SeqC Aggiunta della coppia C al precedente allineamento DGEKFGPPQRSGQRSG |||.||| ||||.||| DGERFGP-QRSGNRSG | |.||. .|||.||| DHEKFGSSQRSGQRSG SeqA SeqB SeqC Allineamento multiplo Per allineare 3 sequenze A, B e C si può procedere per passi successivi. Prima si allineano A e B e in seguito la sequenza C viene aggiunta all’allineamento. Dal momento che i risultati sono differenti a seconda dell’ordine di aggiunta delle sequenze, si provano prima tutti gli allineamenti a coppie e quindi si procede allineando via via le sequenze più vicine. In questo modo vengono minimizzati gli errori, dal momento che si commettono in genere errori maggiori nell’allineare sequenze più divergenti. Questi errori sarebbero propagati agli allineamenti successivi Biochimica computazionale

22
Allineamento progressivo
Allineamento multiplo Allineamento progressivo Allineamenti a coppie [N(N-1)/2 confronti] di tutte le sequenze con programmazione dinamica o metodi approssimati (BLAST, FASTA). Calcolare una matrice diagonale di distanze. Costruire un albero (Neighbor-joining, UPGMA, etc.) sulla base della matrice. L’albero serve da guida per gli allineamenti successivi. Cominciando dal primo nodo aggiunto all’albero, che rappresenta le due sequenze più vicine, allineare via via gli altri nodi (i quali possono essere due sequenze, una sequenza ed un allineamento o due allineamenti) fino a che tutte le sequenze sono state allineate Biochimica computazionale
/2 confronti] di tutte le sequenze con programmazione dinamica o metodi approssimati (BLAST, FASTA). Calcolare una matrice diagonale di distanze. Costruire un albero (Neighbor-joining, UPGMA, etc.) sulla base della matrice. L’albero serve da guida per gli allineamenti successivi. Cominciando dal primo nodo aggiunto all’albero, che rappresenta le due sequenze più vicine, allineare via via gli altri nodi (i quali possono essere due sequenze, una sequenza ed un allineamento o due allineamenti) fino a che tutte le sequenze sono state allineate. Biochimica computazionale.")
23
Feng-Doolittle algorithm
Does all pairwise alignments and scores them Converts pairwise scores to “distances” D = -log Seff = -log [(Sobs –Srand)/(Smax –Srand)] Sobs = pairwise alignment score Srand = expected score for random alignment Smax = average of self-alignments of the two sequences
/(Smax –Srand)] Sobs = pairwise alignment score. Srand = expected score for random alignment. Smax = average of self-alignments of the two sequences.")
24
L’albero guida e la clusterizzazione
2° 1 Hbb_human Hbb_horse Hba_human Hba_horse Myg_whale b_hu b_ho a_hu a_ho M_w E’ una matrice di distanze, minore è il numero, maggiore è la similitudine... 1° 2° Ordine di clusterizzazione 3° 4° 1° PEEKSAVTALWGKVN--VDEVGG Hbb_human GEEKAAVLALWDKVN--EEEVGG Hbb_horse PADKTNVKAAWGKVGAHAGEYGA Hba_human AADKTNVKAAWSKVGGHAGEYGA Hba_horse EHEWQLVLHVWAKVEAGVAGHGQ Myg_whale Allineamento finale

25
Allineamento progressivo: CLUSTAL
Higgins & Sharp 1988 Matrice di distanza ottenuta con confronti a coppie Albero filogenetico di neighbor-joining costruito dalla matrice Allineamento progressivo delle coppie (sequenza-sequenza, sequenza-profilo, profilo -profilo) utilizzando l’albero come guida
 utilizzando l’albero come guida.")
26
CLUSTALW improvement Thompson et al 1994 Le sequenze filogeneticamente più distanti ricevono un peso proporzionalmente più alto nell’allineamento La penalità da assegnare ai gap dipende dal tipo di residui come osservato in sequenze a struttura nota (Pascarella & Argos) La penalità dipende anche dalla posizione. Se ci sono gap nelle vicinanze la penalità aumenta
 La penalità dipende anche dalla posizione. Se ci sono gap nelle vicinanze la penalità aumenta.")
27
CLUSTALW e CLUSTALX Allineamento delle globine ottenuto con CLUSTALW
CLUSTALX. Stesso algoritmo con interfaccia grafica (PC)
")
28
Uno score così è chiamato Objective Function (OF)
Valutare la bontà di un multi-allineamento In genere: si sommano tutti gli score di tutte le possibili coppie di proteine allineate, pesando i valori in base alla similitudine nello stesso cluster per evitare che alcuni cluster prevalgano su altri nel conteggio finale. Ottengo un WSP (Weighted Sum of Pairs): WSPscore = Σ Σ Wij Ŝ(Aij) i = j = 1 N N N: numero di sequenze i,j: coppia di sequenze Ŝ: punteggio di similarità della coppia W: peso per la coppia Il valore complessivo del WSP dipende dai criteri di punteggio utilizzati nell’allineamento più che da considerazioni biologiche, ma è comunque un criterio valido per tutti gli allineamenti con gli stessi parametri Uno score così è chiamato Objective Function (OF)
: WSPscore = Σ Σ Wij Ŝ(Aij) i = 1 j = 1. N-1 N. N: numero di sequenze i,j: coppia di sequenze Ŝ: punteggio di similarità della coppia W: peso per la coppia. Il valore complessivo del WSP dipende dai criteri di punteggio utilizzati nell’allineamento più che da considerazioni biologiche, ma è comunque un criterio valido per tutti gli allineamenti con gli stessi parametri. Uno score così è chiamato Objective Function (OF)")
29
Allineamento iterativo
Il primo allineamento multiplo viene usato per predire un nuovo albero, nuovi pesi e nuovi allineamenti fino a che non si ha un miglioramento nel punteggio dell’allineamento. Implementato in PRRP, DIALIGN

30
Comparazione dei metodi di allineamento multiplo
Thompson et al 1999 Set di riferimento BAliBASE Una bancadati di proteine allineate strutturalmente e suddivisa in set che rappresentano problemi tipici che si hanno in allineamenti multipli: - estensioni terminali - inserzioni - famiglia rispetto ad orfani

31
Comparazione dei metodi di allineamento multiplo
Colonne correttamente allineate V1= <25% id. V2= 20-40% id. V3= >35% id. Sequenze di lunghezza simile Orfani allineati ad una famiglia Sequenze con estensioni terminali Conclusioni: 1) per sequenze di lunghezza comparabile i sistemi globali e iterativi funzionano meglio; 2) per allineare una sequenza orfana ad una famiglia conviene usare i sistemi progressivi (CLUSTALX) e si ottengono risultati migliori se si usano molti membri della famiglia; 3) se le sequenze presentano diverse estensioni alle estremità N e C terminali conviene utilizzare sistemi di allineamento locale
 per sequenze di lunghezza comparabile i sistemi globali e iterativi funzionano meglio; 2) per allineare una sequenza orfana ad una famiglia conviene usare i sistemi progressivi (CLUSTALX) e si ottengono risultati migliori se si usano molti membri della famiglia; 3) se le sequenze presentano diverse estensioni alle estremità N e C terminali conviene utilizzare sistemi di allineamento locale.")
32
Utilizzo dei colori I file raw-text possono essere utilizzati per visualizzare le colonne, ma è possibile associare colori diversi per residui con caratteristiche chimico fisiche diverse. Questo facilita molto la visualizzazione dei multiallineamenti

33
Rappresentazioni dell’allineamento multiplo: conservazione

34
Rappresentazioni dell’allineamento multiplo: sostituzioni

35
Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica a nessuna delle proteine del dataset. si possono definire dei simboli che la definiscano e che indichino anche conservazioni non perfette in una posizione. è possibile utilizzare una formattazione precisa che permetta di capire anche le variazioni in una posizione, non solo le conservazioni.

36
Alcuni modi di indicare le sequenze consenso
Consenso esatto Consenso a simboli Consenso con variazioni GRVQGV--R------A--LG—-GWV GRVQGh-aRvvvvvvAvvLGivGWV GRVQG[VI]-[FY]R------A—L----GWY GRVQGV--R-6A—LG--GWV Consenso con ripetizioni

37
Profili dei multi-allineamenti
Un multi-allineamento genera molte più informazioni per l’individuazione dei residui importanti per una famiglia di proteine di tanti allineamenti a coppie. Diventa quindi basilare poter riassumere le conservazioni osservate in un unico formato. Inoltre multi-allineare proteine divergenti tra loro è molto più informativo rispetto alla stessa analisi fatta su proteine molto simili. Un PROFILO è un metodo di SCORING in cui ad ognuno dei venti amino acidi viene assegnato un punteggio basato sulla frequenza e sul valore in una matrice di sostituzione. Ogni cella di un profilo esprime quindi il peso da attribuire ad ogni aminoacido in quella posizione

38
Profili Gribskov et al 1987 Penalità allungamento gap
HBA_HUMAN ...vga--hagey... HBB_HUMAN ...v----nvdev... MYG_PHYCA ...vea--dvag-... GLB3_CHITP ...vkg------d... GLB5_PETMA ...vys--tyets... LGB2_LUPLU ...fna--nipkh... GLB1_GLYDI ...iagadngagv... Gribskov et al 1987 Penalità allungamento gap Sequenza di consenso dell’allineamento Penalità apertura gap Un profilo rappresenta l’informazione di un allineamento multiplo assegnando a ciascuna colonna dell’allineamento punteggi specifici per ciascun amino acido e per i gap. E’ rappresentato in figura un profilo ottenuto con una porzione allineata di alcune globine. In ciascuna riga è rappresentato l’amino acido di consenso ed i valori di punteggio per i vari amino acidi della corrispondente colonna dell’allineamento (la prima riga corrisponde alla prima colonna dell’allineamento e così via). Gli amino acidi che hanno punteggio maggiore (sottolineati) sono indicati nel consenso.
. Gli amino acidi che hanno punteggio maggiore (sottolineati) sono indicati nel consenso.")
39
Calcolo dei punteggi nei profili
HBA_HUMAN ...vga--hagey... HBB_HUMAN ...v----nvdev... MYG_PHYCA ...vea--dvag-... GLB3_CHITP ...vkg------d... GLB5_PETMA ...vys--tyets... LGB2_LUPLU ...fna--nipkh... GLB1_GLYDI ...iagadngagv... Punteggio di un aminoacido X per la prima colonna: Dove s(Y,X) è il punteggio della sostituzione dell’aminoacido Y in X nelle matrici PAM o BLOSUM. I punteggi per i vari amino acidi nelle colonne sono calcolati dalle matrici di sostituzione assegnando un “peso” diverso ai punteggi a seconda della frequenza degli amino acidi nell’allineamento. Non è un modello probabilisticamente rigoroso e produce alcune anomalie.
 è il punteggio della sostituzione dell’aminoacido Y in X nelle matrici PAM o BLOSUM. I punteggi per i vari amino acidi nelle colonne sono calcolati dalle matrici di sostituzione assegnando un peso diverso ai punteggi a seconda della frequenza degli amino acidi nell’allineamento. Non è un modello probabilisticamente rigoroso e produce alcune anomalie.")
40
Sequence logos Rappresentazione grafica del grado di conservazione delle colonne dell’allineamento. Derivano dalla teoria dell’informazione di Shannon: Massima incertezza di osservare uno su M simboli equiprobabili H = log2(M) = - log2(P) Quando i simboli non sono equiprobabili si utilizza la formula generale di Shannon Nel grafico è rappresentata in ordinata la conservazione come diminuzione di incertezza. Nel caso dei nucleotidi log2(4) - H. Per i gli aminoacidi log2(20) - H La massima conservazione in bits è quindi 2 per i nucleotidi e 4.32 per gli amino acidi.
 = - log2(P) Quando i simboli non sono equiprobabili si utilizza la formula generale di Shannon. Nel grafico è rappresentata in ordinata la conservazione come diminuzione di incertezza. Nel caso dei nucleotidi log2(4) - H. Per i gli aminoacidi log2(20) - H. La massima conservazione in bits è quindi 2 per i nucleotidi e 4.32 per gli amino acidi.")
41
Hidden Markov Models (HMM)
Krog, Haussler Eddy, Durbin transizioni stato Gli “Hidden Markov Models” (HMM) sono una classe di modelli probabilistici che si applicano a serie temporali o sequenze lineari. Un modello è caratterizzato da una determinata architettura composta da stati e da transizioni di stato. Ciascuno stato ha una data probabilità di emettere simboli o di effettuare una transizione. Il modello in figura rappresenta sequenze composte da due lettere (a,b) generate da due stati, ciascuno con una diversa probabilità di emissione. Partendo dalla stato 1 il modello ha generato una sequenza (aba) attraverso una successione di stati. Ciò che viene osservata è la sequenza di simboli, mentre la successione degli stati rimane nascosta (hidden). La probabilità combinata P(x, | HMM) della sequenza di simboli osservata è il prodotto di tutte le probabilità di emissioni e transizioni.
 sono una classe di modelli probabilistici che si applicano a serie temporali o sequenze lineari. Un modello è caratterizzato da una determinata architettura composta da stati e da transizioni di stato. Ciascuno stato ha una data probabilità di emettere simboli o di effettuare una transizione. Il modello in figura rappresenta sequenze composte da due lettere (a,b) generate da due stati, ciascuno con una diversa probabilità di emissione. Partendo dalla stato 1 il modello ha generato una sequenza (aba) attraverso una successione di stati. Ciò che viene osservata è la sequenza di simboli, mentre la successione degli stati rimane nascosta (hidden). La probabilità combinata P(x, | HMM) della sequenza di simboli osservata è il prodotto di tutte le probabilità di emissioni e transizioni.")
42
Profili HMM inserzione match delezione
Gli HMM possono essere usati per modellare allineamenti multipli di sequenze di una data famiglia (profili HMM). L’architettura del modello prevede tre tipi di stati ( match, inserzione, delezione), più uno stato finale ed uno stato iniziale. L’HMM rappresentato in figura modella l’allineamento multiplo delle sei sequenze (3 colonne) visualizzato a fianco. La probabilità di emissione dei vari aminoacidi derivano dalle frequenze osservate nell’allineamento. Modelli generati con allineamento multipli possono essere impiegati per: 1) allineare nuove sequenze al modello; 2) individuare in un database sequenze aderenti al modello; 3) individuare corrispondenze tra una sequenza e un database di modelli.
. L’architettura del modello prevede tre tipi di stati ( match, inserzione, delezione), più uno stato finale ed uno stato iniziale. L’HMM rappresentato in figura modella l’allineamento multiplo delle sei sequenze (3 colonne) visualizzato a fianco. La probabilità di emissione dei vari aminoacidi derivano dalle frequenze osservate nell’allineamento. Modelli generati con allineamento multipli possono essere impiegati per: 1) allineare nuove sequenze al modello; 2) individuare in un database sequenze aderenti al modello; 3) individuare corrispondenze tra una sequenza e un database di modelli.")
43
Profili HMM: calcolo parametri
seq xxx--xxxxx... seq x----xxxxx... seq xxx--xxxx-... seq xxx------x... seq xxx--xxxxx... seq xxx--xxxxx... Seq xxxxxxxxxx... m1 m2 m8 D1 BEGIN END I0 D2 I1 tM1I1 tM1M2 tM1D2 Il modello è costruito in modo da assegnare tanti stati “match” quante sono le colonne dell’allineamento in cui gli aminoacidi prevalgono sui gap. I parametri iniziali dipendono dal numero di simboli emessi e dal numero di transizioni (frecce che partono da uno stato). In questo modo, prima di leggere un allineamento le probabilità di emissione di ciascun amino acido dello stato m1 saranno eM1(x) = 1/20, Le probabilità di transizione dallo stato m1 saranno tM1M2=1/3, tM1D2=1/3, tM1I1 =1/3. I parametri vengono quindi modificati in base all’allineamento in ragione delle occorrenze dei caratteri in una colonna e le occorrenze delle transizioni.
. In questo modo, prima di leggere un allineamento le probabilità di emissione di ciascun amino acido dello stato m1 saranno eM1(x) = 1/20, Le probabilità di transizione dallo stato m1 saranno tM1M2=1/3, tM1D2=1/3, tM1I1 =1/3. I parametri vengono quindi modificati in base all’allineamento in ragione delle occorrenze dei caratteri in una colonna e le occorrenze delle transizioni.")
44
Profili HMM: pseudocounts
HBA_HUMAN ...vga--hagey... HBB_HUMAN ...v----nvdev... MYG_PHYCA ...vea--dvag-... GLB3_CHITP ...vkg------d... GLB5_PETMA ...vys--tyets... LGB2_LUPLU ...fna--nipkh... GLB1_GLYDI ...iagadngagv... m1 m2 m8 D1 BEGIN END I0 D2 I1 Quando una transizione o l’emissione di un particolare simbolo non si osserva è necessario aggiungere finti conti (“pseudocounts”) per evitare valori di probabilità zero. Il sistema più semplice di pseudocounts è la regola di Laplace: aggiungere 1 a tutte le frequenze. Usando questo sistema nella prima colonna occorre aggiungere 17 conti per gli aminoacidi “mancanti”. Il numero totale sarà quindi (5+1)v+(1+1)f+(1+1)i+(0+17)x = 27. Le probabilità di emissione saranno em1(V)= 6/27, em1(I)=2/27, em1(F)=2/27, em1(x)=1/27. Analogamente per le probabilità di transizione, in cui osserviamo 6 transizioni allo stato di match, 1 allo stato di delezione e zero allo stato di inserzione: tm1m2=7/9, tm1d2=2/9, tm1I1= 1/10. Questo sistema di pseudocount funziona solo quando il campione di sequenze è sufficientemente numeroso (>50); in altri casi vanno usati sistemi più sofisticati (misture di Dirichlet, misture basate sulle matrici di sostituzione, stime basate sull’ancestore).
 per evitare valori di probabilità zero. Il sistema più semplice di pseudocounts è la regola di Laplace: aggiungere 1 a tutte le frequenze. Usando questo sistema nella prima colonna occorre aggiungere 17 conti per gli aminoacidi mancanti . Il numero totale sarà quindi (5+1)v+(1+1)f+(1+1)i+(0+17)x = 27. Le probabilità di emissione saranno em1(V)= 6/27, em1(I)=2/27, em1(F)=2/27, em1(x)=1/27. Analogamente per le probabilità di transizione, in cui osserviamo 6 transizioni allo stato di match, 1 allo stato di delezione e zero allo stato di inserzione: tm1m2=7/9, tm1d2=2/9, tm1I1= 1/10. Questo sistema di pseudocount funziona solo quando il campione di sequenze è sufficientemente numeroso (>50); in altri casi vanno usati sistemi più sofisticati (misture di Dirichlet, misture basate sulle matrici di sostituzione, stime basate sull’ancestore).")
45
Profili HMM: weighting
I parametri dell’HMM vanno stimati assegnando un peso minore all’informazione portata da sequenze simili ed un peso maggiore all’informazione portata da sequenze più divergenti. Vi sono metodi di weighting basati su alberi filogenetici ed altri indipendenti da questi (Voroni, Maximum discrimination, Maximum Entropy). Tutti i sistemi di weighting soffrono di un problema comune. Se in un modello di una famiglia si inserisce una sequenza errata (non appartenente alla famiglia) questa avrà in proporzione molto peso sul modello finale. V In un intuitivo sistema di weighting si immagina di applicare un voltaggio alla radice dell’albero e di pesare le sequenze in proporzione alla corrente che arriva a ciascun nodo.
. Tutti i sistemi di weighting soffrono di un problema comune. Se in un modello di una famiglia si inserisce una sequenza errata (non appartenente alla famiglia) questa avrà in proporzione molto peso sul modello finale. V. In un intuitivo sistema di weighting si immagina di applicare un voltaggio alla radice dell’albero e di pesare le sequenze in proporzione alla corrente che arriva a ciascun nodo.")
46
Questioni legate ai profili HMM
Dato un allineamento, quali sono la struttura e i parametri di un HMM che descrivono nel modo migliore possibile la conservazione (APPRENDIMENTO)? Qual è la sequenza ottimale di stati con cui un HMM genera una data sequenza (ALLINEAMENTO)? Qual è la probabilita che una data sequenza sia stata generata da un HMM (SIGNIFICATIVITA’)?
 Qual è la sequenza ottimale di stati con cui un HMM genera una data sequenza (ALLINEAMENTO) Qual è la probabilita che una data sequenza sia stata generata da un HMM (SIGNIFICATIVITA’)")
47
Hmmer Sean Eddy Hmmer (pronunciato “hammer”) è un pacchetto di programmi per creare ed usare modelli markoviani di allineamenti di DNA e proteine. I programmi principali sono: Hmmalign: allineare sequenze ad un HMM Hmmbuild: creare un HMM a partire da un allineamento Hmmsearch: cercare un database di sequenze con un HMM
 è un pacchetto di programmi per creare ed usare modelli markoviani di allineamenti di DNA e proteine. I programmi principali sono: Hmmalign: allineare sequenze ad un HMM. Hmmbuild: creare un HMM a partire da un allineamento. Hmmsearch: cercare un database di sequenze con un HMM.")
48
Protein family PFAM (acronimo di Protein Families) è un database di domini di proteine descritti con modelli markoviani. E’ diviso in due sezioni: pfam-A contiene allineamenti curati da esperti; pfam-B contiene sequenze che vengono automaticamente raggruppate.
 è un database di domini di proteine descritti con modelli markoviani. E’ diviso in due sezioni: pfam-A contiene allineamenti curati da esperti; pfam-B contiene sequenze che vengono automaticamente raggruppate.")
49
Pfam

50
Pfam: family description

51
Ricerca di omologia in Pfam: “anatomia” delle proteine

Presentazioni simili



















>")

