Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
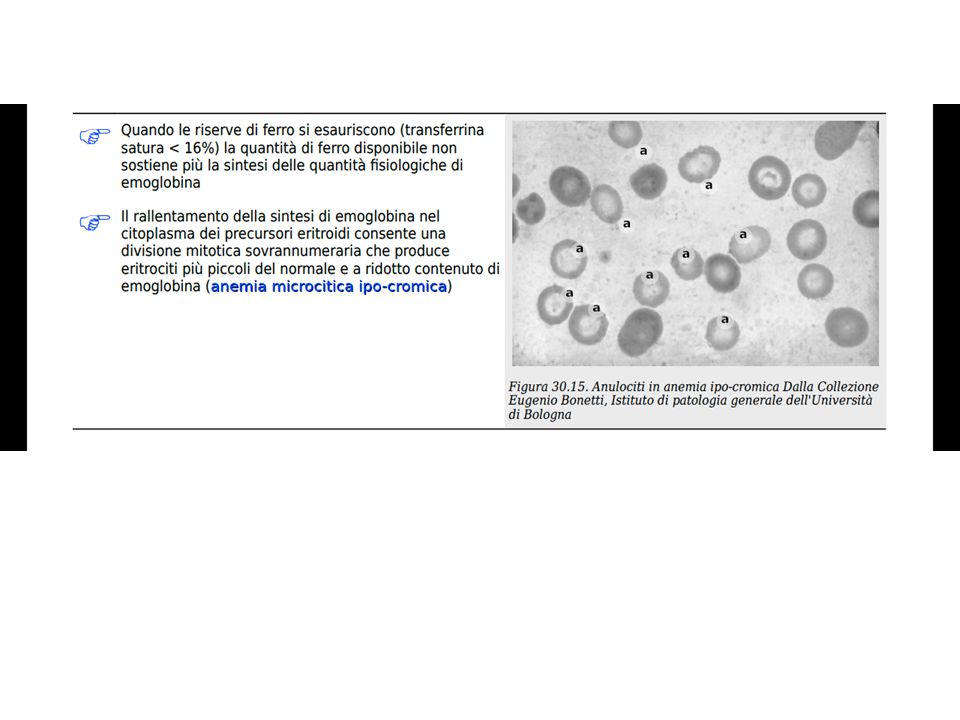
Anemie da disordini della maturazione

2
Disordini della maturazione
The low reticulocite production index is a reflection of the ineffective erithropoiesis that results from the destruction within the marrow of the developing eritroblasts. Marrow morfology shows M/E ratio < 1:1, diagnostic of eritroid iperplasia. Difetti maturazione a livello citoplasmatico: Microcitosi e ipocromia da Hb Sideropenia grave Anemia sideropenica Anemia sideroblastica Talassemia (difetti produzione di emoglobina) Difetti maturazione a livello nucleare: macrocitosi e sviluppo anomalo del midollo Deficienza di folati Def. Vitamina B12 Tossicità da farmaci (MTX, agenti alchilanti) che interferiscono con sintesi DNA Anemia refrattaria (mielodisplasia) Maturation disorders The presence of anemia with an inappropriately low reticulocite production index, macro- or microcitosis on smear, and abnormal red cell indices suggests a maturation disorder. Maturation disorders are divided into two categories: nuclear maturation defects, associated with macrocitosis and abnormal marrow development, and citoplasmic maturation defects, associated with microcitosis and hypochromia usually from defects in hemoglobin synthesis. The low reticulocite production index is a reflection of the ineffective erithropoiesis that results from the destruction within the marrow of the developing eritroblasts. Marrow morfology shows am M/E ratio < 1:1, diagnostic of eritroid iperplasia. Nuclear maturation defects result from vitamin B12 or folic acid deficiency, drug damage, or mielodisplasia. Drugs that interfere with cellular DNA metabolism, such as metotrexate or alkilating agents can produce a nuclear maturation defect. Alcool alone is also capable of producing macrocitosis and a variable degree of anemia, but this is usually associated with coincident folic acid deficiency. Measurements of folic acid and vitamin B12 are key not only in identifying the specific vitamin deficiency but also because they reflect different pathogenetic mechanisms. Cytoplasmic maturation defects result from severe iron deficiency or abnormalities in globin or heme synthesis.
 Difetti maturazione a livello nucleare: macrocitosi e sviluppo anomalo del midollo. Deficienza di folati. Def. Vitamina B12. Tossicità da farmaci (MTX, agenti alchilanti) che interferiscono con sintesi DNA. Anemia refrattaria (mielodisplasia) Maturation disorders. The presence of anemia with an inappropriately low reticulocite production index, macro- or microcitosis on smear, and abnormal red cell indices suggests a maturation disorder. Maturation disorders are divided into two categories: nuclear maturation defects, associated with macrocitosis and abnormal marrow development, and citoplasmic maturation defects, associated with microcitosis and hypochromia usually from defects in hemoglobin synthesis. The low reticulocite production index is a reflection of the ineffective erithropoiesis that results from the destruction within the marrow of the developing eritroblasts. Marrow morfology shows am M/E ratio < 1:1, diagnostic of eritroid iperplasia. Nuclear maturation defects result from vitamin B12 or folic acid deficiency, drug damage, or mielodisplasia. Drugs that interfere with cellular DNA metabolism, such as metotrexate or alkilating agents can produce a nuclear maturation defect. Alcool alone is also capable of producing macrocitosis and a variable degree of anemia, but this is usually associated with coincident folic acid deficiency. Measurements of folic acid and vitamin B12 are key not only in identifying the specific vitamin deficiency but also because they reflect different pathogenetic mechanisms. Cytoplasmic maturation defects result from severe iron deficiency or abnormalities in globin or heme synthesis.")
3
Anemia sideropenica e anemia sideroblastica
If the iron deficiency anemia is mild to moderate, eritroid marrow proliferation is decreased and the anemia is classified as hypoproliferative. IF the anemia is severe and prolonged, the eritroid marrow will become hyperplastic despite the inadeguate iron supply, and the anemia will be classified as ineffective eritropoiesis with a citoplasmic maturation defect. In ambedue i casi l’indice di produttività dei reticolociti è basso Un tipo di anemia sideropenica è quella in cui la mancata formazione dell’eme è legata ad accumulo del ferro nei mitocondri dei sideroblasti midollari che assumono la forma di sideroblasti ad anello. L’ anemia sideroblastica è conseguenza di mielodisplasia. Iron deficiency occupies an unusual position in the classification of anemia. If the iron deficiency anemia is mild to moderate, erutroid marrow proliferation is decreased and the anemia is classified as hypoproliferative. However is the anemia is severe and prolonged, the eritroid marrow will become hyperplastic despite the inadeguate iron supply, and the anemia will be classified as ineffective eritropoiesis with a citoplasmic maturation defect. In either case, a reduced reticulocyte production index, microcitosis, and a classic pattern of iron values make the diagnosis clear and easily distinguish iron deficiency from other cytoplasmic maturation defects such as the thalassemias. Defects in heme sintesis, in contrast to globin sintesis are less common and may be acquired or inherited. Acquired abnormalities are usually associated with mielodisplasia, may lead to either a macro- or microcitic anemia and are frequently associated with mitocondrial iron loading. In these cases, iron is taken up by the mitocondria of the developing eritroid cell but not incorporated into heme. The iron-encrusted mithocondria surround the nucleus of the eritroid cell forming a ring. Based on the distinctive finding of so called ring sideroblasts on the marrow iron stain, patients are diagnosed as having a sideroblastic anemia - almost always reflecting mielodisplasia. Again, studies of iron parameters are helpful in the differential diagnosis and management of these patients.

4
Processi infettivi intercorrenti possono complicare il quadro della anemia sideropenica

6
Anemie da disordini della maturazione

8
Classificazione dell’emoglobinopatie e talassemie:
alterazioni strutturali dell’Hb: anomala polimerizzazione Hb (HbS) anomala cristallizzazione Hb (HbC) emoglobine instabili emoglobine con affinita’ per l’O2 (policitemia); emoglobine con affinita’ per l’O2 (cianosi); difetti quantitativi nella produzione delle catene globiniche -talassemia -talassemia -talassemia, -talassemia, -talasemia
 anomala cristallizzazione Hb (HbC) emoglobine instabili. emoglobine con affinita’ per l’O2 (policitemia); emoglobine con affinita’ per l’O2 (cianosi); difetti quantitativi nella produzione delle catene globiniche. -talassemia. -talassemia. -talassemia, -talassemia, -talasemia.")
9
eme emoglobina = tetramero di 4 catene globiniche + 4 gruppi eme
Ferro++ + protoporfirina IX eme

11
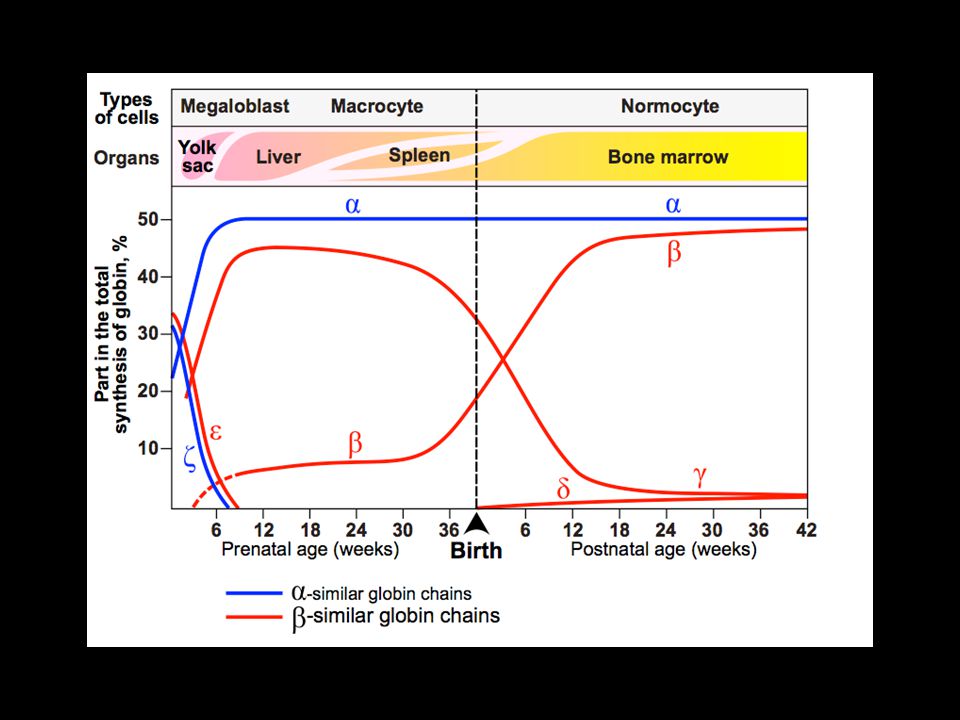
nascita 3 mesi 6 mesi prenatale HbA: 22 95%
HbA1c: 22(glic) 3% HbA2: 2 % HbF: 2 <1% Gower 1 22 Gower 2 22 Portland 22 HbH 4 Hb Barts 4
 3% HbA2: 22 2% HbF: 22 <1% Gower 1 22. Gower 2 22. Portland 22. HbH 4. Hb Barts 4.")
12
Alfa-talassemia Fenotipi clinici di diversa gravità in relazione alla attività residua dell’allele normale

13
-talassemia = difetto produzione catene globiniche
eccesso produzione catene globiniche

14
Regione regolatoria Emoglobina Z
Many different a-thalassemia-1 deletions have been described. The southeast Asian (--SEA) and Filipino --FIL) are most common in US immigrant population. --RA is interesting in that thalassemia results from deletion of a regulatory element (HS-40) rather than the structural a-globin genes. Deletions that spare the z-globin gene can result in increased z-globin expression in newborns and adults. Regione regolatoria Emoglobina Z
 and Filipino --FIL) are most common in US immigrant population. --RA is interesting in that thalassemia results from deletion of a regulatory element (HS-40) rather than the structural a-globin genes. Deletions that spare the z-globin gene can result in increased z-globin expression in newborns and adults. Regione regolatoria. Emoglobina Z.")
15
Base genetica di a thalassemia: sopratutto delezioni
chr. 16 Neither release O2 a2 a1 As I said before, the genetic basis of alpha thalassemias is mostly deletions: If you have 4 functional alpha genes, you are normal. With 3, you are a silent carrier. With 2 you have alpha thalassemia trait which is clinically benign but there is mild microcytic anemia. With only one alpha chain, you have severe hemolytic anemia with primarily HbH, composed of 4 beta chains. This is clinically severe. In the absence of alpha chain in the fetus, the gamma forms a tetramer and is called Hb Bart’s. B4 tetramer is called Hb H. Neither of these hemoglobins is capable of releasing oxygen to the tissues. Infants with severe alpha Thalassemia and Hb Barts suffer severe intrauterine hypoxia and are born with massive generalized fluid accumulation,a condition known as hydrops fetalis. Per capire come si formano Hb H e Hb Barts dobbiamo ricorrere allo schema della ontogenesi della emoglobina e ricordare che le catene gamma sono operative principalmente nella vita fetale mentre le catene beta diventano disponibili solo a partire dalla nascita. Anemia grave clinicamente severa Hb H (b4) Lieve anemia microcitica clinicamente benigna hemoglobin normale Portatore silente Hb Barts (g4) hydrops fetalis Tratto talassemico
 Lieve anemia microcitica clinicamente benigna. hemoglobin normale. Portatore silente. Hb Barts (g4) hydrops fetalis. Tratto talassemico.")
17
Beta- talassemia 17

18
X X X X tratto -talassemico tipo 2 portatore sano omozigote
eterozigote X tratto -talassemico tipo 1 X malattia da HbH X idrope fetale

19
eccesso catene ridotta quantita Hb per RBC (ipocromia) ridotta produzione RBC maturi (iporigenerazione) ridotta sopravvivenza RBC Ridotta produzione di RBC fragili anisopoichilocitosi sequestrazione splenica splenomegalia ipersplenismo anemia aumentato assorbimento Fe difettoso utilizzo Fe accumulo Fe emocromatosi trasfusioni ittero calcoli biliari cirrosi endocrinopatie cardiomiopatia

20
(homozygous a-Thalassemia)
Hydrope fetalis (homozygous a-Thalassemia) Individui con alpha Thalassemia grave e Hb Barts soffrono di grave ipossia intrauterine e nascono con massiccio accumulo di liquidi When two a-thalassemia-1 alleles are inherited, the offspring have no functional a-globin genes, which is not compatible with life. The clinical syndrome is referred to as hemoglobin Bart’s hydrops fetalis, referring to the hemoglobin Bart’s (g4) seen in cord blood. This occurs in same populations as a-thalassemia-1. The clinical laboratory can be helpful in preventing occurrence of Hb Bart’s hydrops fetalis through testing of prospective parents and prenatal diagnosis. Perché questo massiccio accumulo di liquido nel tessuto extra-vascolare ?
 Individui con alpha Thalassemia grave e Hb Barts soffrono di grave ipossia intrauterine e nascono con massiccio accumulo di liquidi. When two a-thalassemia-1 alleles are inherited, the offspring have no functional a-globin genes, which is not compatible with life. The clinical syndrome is referred to as hemoglobin Bart’s hydrops fetalis, referring to the hemoglobin Bart’s (g4) seen in cord blood. This occurs in same populations as a-thalassemia-1. The clinical laboratory can be helpful in preventing occurrence of Hb Bart’s hydrops fetalis through testing of prospective parents and prenatal diagnosis. Perché questo massiccio accumulo di liquido nel tessuto extra-vascolare")
21
Decreased Increased Increased fluid efflux from intravascular space
Pressione osm coll Pressione idrost vasc Increased fluid efflux from intravascular space Capillary leak

22
Pathogenesis of Hydrops
Severe anemia Hepatic extramedullary hematopoeisis Decreased prdtn of plasma proteins Decreased plasma COP COP = colloidal osmotic pressure

23
Pathogenesis of Hydrops
Congestive heart failure Increased central venous pressure Increased capillary hydrostatic pressure

24
Pathogenesis of Hydrops
Severe tissue hypoxia Endothelial cell damage Capillary leak of fluid & protein

25
The Fetal Microcirculation
Adult Sheep 1 l NS bolus IV 30% intravascular at 30 min Fetal Sheep 1 l NS bolus IV 6% intravascular at 30 min Aumento nel feto della permeabilità capillare e della capacità assorbimento spazio interstiziale

26
Morbo di Cooley - talassemia: difetto produzione catene globiniche
HbA HbF HbA2 eccesso catene globiniche - talassemia: difetto produzione catene globiniche

27
eccesso catene precipitazione corpi inclusi nei progenitori eritroidi eritropoiesi inefficace a livello midollare ridotta quantita Hb per RBC (ipocromia) ridotta produzione RBC maturi (iporigenerazione) ridotta sopravvivenza RBC maturazione di pochi RBC difettosi anisopoichilocitosi sequestrazione splenica splenomegalia ipersplenismo anemia ipossia tessutale iperproduzione Epo espansione emopoiesi deformita’ ossea fratture emopoiesi extramidollare deficit folati aumentato assorbimento Fe difettoso utilizzo Fe accumulo Fe emocromatosi trasfusioni ittero calcoli biliari cirrosi endocrinopatie cardiomiopatia
 ridotta produzione RBC maturi. (iporigenerazione) ridotta sopravvivenza RBC. maturazione. di pochi RBC difettosi. anisopoichilocitosi. sequestrazione. splenica. splenomegalia. ipersplenismo. anemia. ipossia tessutale. iperproduzione Epo. espansione. emopoiesi. deformita’ ossea. fratture. emopoiesi extramidollare. deficit folati. aumentato. assorbimento Fe. difettoso. utilizzo Fe. accumulo Fe. emocromatosi. trasfusioni. ittero. calcoli biliari. cirrosi. endocrinopatie. cardiomiopatia.")
28
Anemie da disordini della maturazione

33
MEGALOBLASTOSI MIDOLLARE

34
MACROCITOSI ERITROCITARIA

35
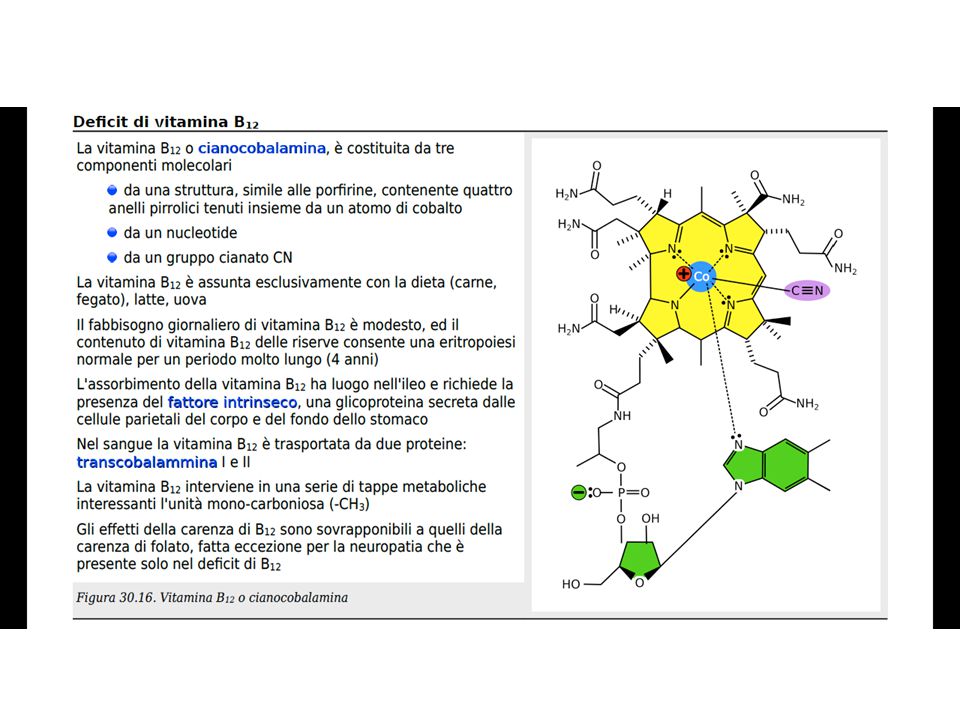
ASSORBIMENTO DELLA VITAMINA B12

36
Co-enzima della METIONINA SINTETASI
ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 METIL-COBALAMINA: Co-enzima della METIONINA SINTETASI METILOMOCISTEINA + N5 TETRAIDROFOLATO METIONINA + TETRAIDROFOLATO (FH4) FH4: Co-enzima della TIMIDILATOSINTETASI URIDINA TIMIDINA FH4 INTERVIENE NELLA SINTESI DELLE PURINE E DI METIONINA* *La metionina è un componente per la sintesi della colina, dei fosfolipidi e della proteina basica della mielina
 FH4: Co-enzima della TIMIDILATOSINTETASI. URIDINA. TIMIDINA. FH4 INTERVIENE NELLA SINTESI DELLE PURINE E DI METIONINA* *La metionina è un componente per la sintesi della colina, dei fosfolipidi e della proteina basica della mielina.")
37
VIT B12 E FOLATI NELLA SINTESI DEL DNA

38
CAUSE DI ANEMIA MEGALOBLASTICA
DEFICIT DI VITAMINA B12 Insufficiente apporto: dieta strettamente vegetale Carenza di FI: anemia perniciosa (Ab anti FI nel 60% casi, anti cell parietali stomaco nel 90% casi) gastrectomia totale/subtotale gastrite atrofica del moncone Malassorbimento intestinale: diverticolosi del tenue ansa cieca parassitosi intestinale sprue tropicale ileite, fistole ileo-coliche, resezione ileale (M. di Crohn)
 gastrectomia totale/subtotale. gastrite atrofica del moncone. Malassorbimento intestinale: diverticolosi del tenue. ansa cieca. parassitosi intestinale. sprue tropicale. ileite, fistole ileo-coliche, resezione ileale (M. di Crohn)")
39
CAUSE DI ANEMIA MEGALOBLASTICA
DEFICIT DI ACIDO FOLICO Insufficiente apporto : ipoalimentazione, diete, alcolismo Malassorbimento intestinale: resezione digiunale enteropatia da glutine sprue tropicale dermatite erpetiforme linfomi intestinali Aumentato consumo: gravidanza, allattamento, nati prematuri Neoplasie aumentato turnover midollare (iperplasia dell’ eritrone) malattie infiammatorie croniche Perdita eccessiva: Dialisi
 malattie infiammatorie croniche. Perdita eccessiva: Dialisi.")
40
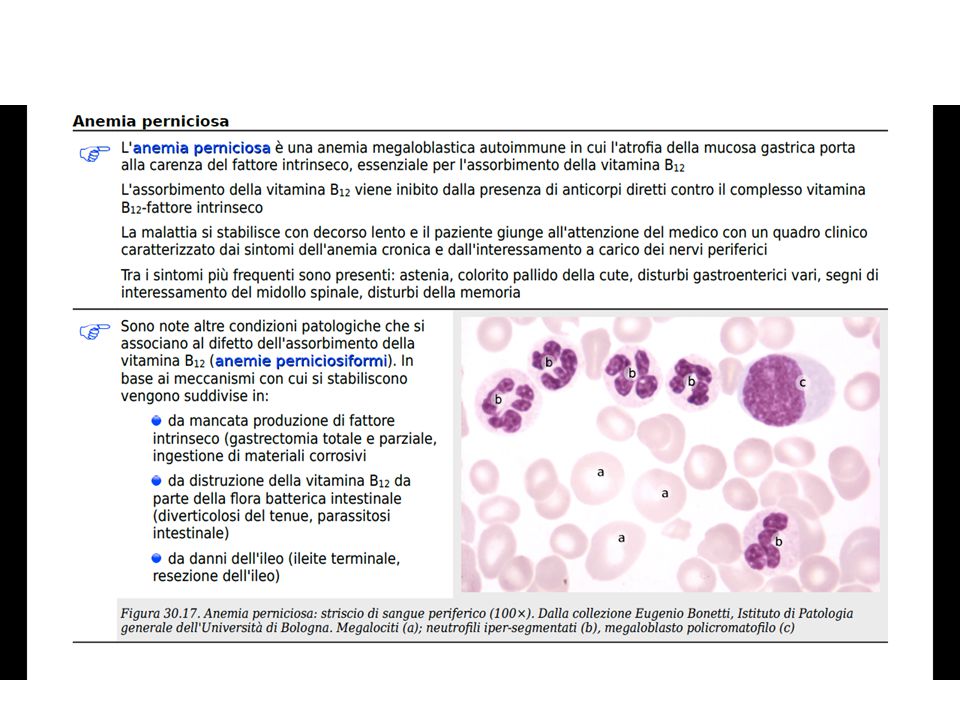
CLASSIFICAZIONE ETIOPATOGENETICA
ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 CLASSIFICAZIONE ETIOPATOGENETICA PERNICIOSA Predisposizione genetica Autoimmune (Ab anti FI mucosa gastrica) PERNICIOSIFORMI Acquisite Non autoimmuni QUADRI CLINICO-LABORATORISTICI SIMILI
 PERNICIOSIFORMI. Acquisite. Non autoimmuni. QUADRI CLINICO-LABORATORISTICI SIMILI.")
41
ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 E FOLATI
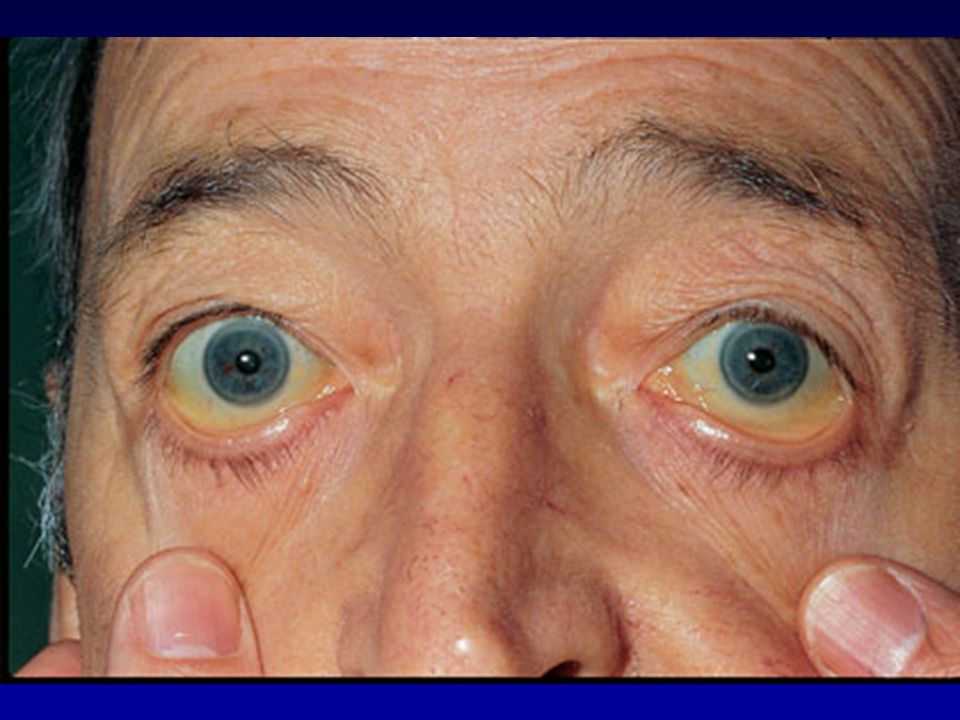
Colorito giallo limone, glossite e cheilite angolare in donna con anemia megaloblastica

42
ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 E FOLATI
Paziente con anemia perniciosa e vitiligine

43
LABORATORIO: CARATTERIZZAZIONE DELL’ANEMIA
ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 E FOLATI LABORATORIO: CARATTERIZZAZIONE DELL’ANEMIA Anemia macrocitica Riduzione reticolociti All’osservazione al microscopio due segni patognomonici: Macrovalocitosi (con anisopoichilocitosi) Ipersegmentazione dei granulociti neutrofili Mielobiopsia: iperplasia dell’eritropoiesi con megaloblastosi e predominanza degli eritroblasti basofili e policromatofili. MKC pseudo-iperdiploidi (ipersegmentati)
 Ipersegmentazione dei granulociti neutrofili. Mielobiopsia: iperplasia dell’eritropoiesi con megaloblastosi e predominanza degli eritroblasti basofili e policromatofili. MKC pseudo-iperdiploidi (ipersegmentati)")
44
EMOCROMO ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 E FOLATI Hb g/dL
NORMALE ANEMIA MEGALOBLASTICA Hb g/dL 15.0 7.5 Eritrociti x 106/L HCT (%) 45 25 MCV (3) 90 125 Leucociti x 103/L 6000 Normali o ridotti Piastrine x 103/L
 MCV (3) Leucociti x 103/L Normali o ridotti. Piastrine x 103/L")
45
LABORATORIO: altri parametri alterati
ANEMIE MEGALOBLASTICHE DA DEFICIT DI VIT B12 E FOLATI LABORATORIO: altri parametri alterati Possibile leucopenia e piastrinopenia Iperbilirubinemia indiretta (bilirubina non coniugata) e aumento di LDH, espressione di aumentata eritroblastolisi endomidollare
 e aumento di LDH, espressione di aumentata eritroblastolisi endomidollare.")
46
Anemie emolitiche

47
Anemia emolitica: aumentata distruzione di RBC nel sangue periferico
assenza di eritropoiesi inefficace a livello midollare iperplasia eritroide a livello midollare emolisi normale tipo congenito (difetto intrinseco) emoglobinopatie (HbS) difetti enzimatici (G6PD, PK) difetti di membrana (sferocitosi) tipo acquisito (difetto estrinseco) traumi meccanici mediate da anticorpi da insulti tossici o fisici
 emoglobinopatie (HbS) difetti enzimatici (G6PD, PK) difetti di membrana (sferocitosi) tipo acquisito (difetto estrinseco) traumi meccanici. mediate da anticorpi. da insulti tossici o fisici.")
48
aumentata distruzione a livello periferico dei RBC per:
Anemia emolitica: aumentata distruzione a livello periferico dei RBC per: emolisi intravascolare emolisi extravascolare INTRAVASCOLARE EXTRAVASCOLARE striscio periferico schistociti sferociti aptoglobina assente/bassa normale/lieve riduzione emoglobina urine assente emosiderina urine assente Coombs diretto negativo positivo LDH aumentata aumentata bilirubina indiretta aumentata aumentata

50
L’ERITROCITO VIVE 120 GIORNI
INVECCHIANDO PERDE CAPACITA’ DI GENERARE ENERGIA E QUINDI DI CONTENERE E RIPARARE I DANNI DA STRESS OSSIDATIVO E DI MANTENERE I LIPIDI DI MEMBRANA. NON E’ IN GRADO DI RINNOVARE LE PROTEINE DELLA MEMBRANA E DEL CITOSCHELETRO. L’ERITROCITO VECCHIO DIVENTA RIGIDO, PERDE PLASTICITA’ E DEFORMABILITA’ E VIENE CATTURATO DAI MACROFAGI: EMOLISI FISIOLOGICA, EXTRAVASCOLARE.

51
EMOLISI EXTRAVASCOLARE (FISIOLOGICA E PATOLOGICA)
L’ERITROCITO “VECCHIO” O “MALATO” VIENE SEQUESTRATO DAI MACROFAGI. EMOLISI INTRAVASCOLARE L’ERITROCITO COLPITO DA UN AGENTE EMOLIZZANTE (PER ES. CON ATTIVAZIONE COMPLETA DELLA CASCATA COMPLEMENTARE) SI “ROMPE” IN CIRCOLO, LIBERANDO EMOGLOBINA (SIERO ROSA, URINE ROSSE NERE)
 SI ROMPE IN CIRCOLO, LIBERANDO EMOGLOBINA (SIERO ROSA, URINE ROSSE NERE)")
52
DOVE AVVIENE L’EMOLISI EXTRAVASCOLARE?
OVUNQUE CI SIANO MACROFAGI. FISIOLOGICAMENTE SOPRATTUTTO NEL MIDOLLO OSSEO, MILZA E FEGATO. IN CONDIZIONI PATOLOGICHE (ERITROCITI DANNEGGIATI, RIVESTITI DA ANTICORPI) LA SEDE PRINCIPALE DELL’EMOLISI DIVENTA LA MILZA, PER LA PARTICOLARE STRUTTURA DELLA POLPA ROSSA. TOGLIERE LA MILZA NON ALLUNGA LA VITA DEGLI ERITROCITI NORMALI, MA PUO’ ALLUNGARE LA VITA DI ERITROCITI ANORMALI.
 LA SEDE PRINCIPALE DELL’EMOLISI DIVENTA LA MILZA, PER LA PARTICOLARE STRUTTURA DELLA POLPA ROSSA. TOGLIERE LA MILZA NON ALLUNGA LA VITA DEGLI ERITROCITI NORMALI, MA PUO’ ALLUNGARE LA VITA DI ERITROCITI ANORMALI.")
53
L’EMOLISI: DALL’EMOGLOBINA ALLA BILIRUBINA
L’ERITROCITO VECCHIO O DANNEGGIATO VIENE FAGOCITATO DAI MACROFAGI. IL FERRO VIENE STACCATO DALL’EME E RIUTILIZZATO (L’EPCIDINA REGOLA LA CAPACITA’ DEI MACROFAGI DI RIMETTERE IN CIRCOLAZIONE IL FERRO). IL NUCLEO TETRAPIRROLICO DELL’EME VIENE APERTO E SI FORMA LA BILIRUBINA (PIGMENTO DI COLORE GIALLO-VERDE).
. IL NUCLEO TETRAPIRROLICO DELL’EME VIENE APERTO E SI FORMA LA BILIRUBINA (PIGMENTO DI COLORE GIALLO-VERDE).")
54
4. LA BILIRUBINA CIRCOLA NEL SANGUE LEGATA ALL’ ALBUMINA.
5. IL COMPLESSO ALBUMINA-BILIRUBINA VIENE RICONOSCIUTO SUL POLO VASCOLARE DEGLI EPATOCITI. LA BILIRUBINA VIENE INTERNALIZZATA. 6. NELL’EPATOCITO LA BILIRUBINA VIENE CONIUGATA DUE VOLTE CON ACIDO GLICURONICO, E DIVENTA IDROSOLUBILE. 7. LA BILIRUBINA DIGLICURONATA VIENE ESCRETA SUL POLO BILIARE DELL’EPATOCITA.

55
8. CANALICOLI BILIARI, DUTTULI BILIARI, DOTTI BILIARI, COLEDOCO (PANCREAS-WIRSUNG), DUODENO.
7. NELL’INTESTINO LA BILIRUBINA SI TRASFORMA IN BILINOGENO E BILINE CHE VENGONO IN GRAN PARTE ELIMINATI CON LE FECI (DONDE IL COLORE SCURO DELLE FECI) E IN PARTE CON LE URINE (DONDE IL COLORITO VARIABILE DELLE URINE, TANTO PIU’ “SCURE” QUANTO MAGGIORE E’ IL LORO CONTENUTO DI UROBILINOGENO).
 E IN PARTE CON LE URINE (DONDE IL COLORITO VARIABILE DELLE URINE, TANTO PIU’ SCURE QUANTO MAGGIORE E’ IL LORO CONTENUTO DI UROBILINOGENO).")
56
11. EMOLISI PATOLOGICA = PIU’ BILIRUBINA = MAGGIOR LAVORO PER GLI EPATOCITI = MAGGIORE ELIMINAZIONE DI BILIRUBINA, BILINOGENI, BILINE = MAGGIOR QUANTITA’ DI UROBILINOGENO NELLE URINE (URINE “FLAMMEE”). 12. SE LA QUANTITA’ DI BILIRUBINA PRODOTTA DALL’EMOLISI ECCEDE LE CAPACITA’ DEL FEGATO, LA CONCENTRAZIONE DELLA BILIRUBINA NON CONIUGATA (INDIRETTA) NEL SANGUE AUMENTA: COLORAZIONE GIALLA CUTE, MUCOSE, CONGIUNTIVE = ITTERO.
 NEL SANGUE AUMENTA: COLORAZIONE GIALLA CUTE, MUCOSE, CONGIUNTIVE = ITTERO.")
57
13. SE LA PROTEINA CHE TRASPORTA LA BILIRUBINA NEGLI EPATOCITI E’ DIFETTIVA = IPERBILIRUBINEMIA NON CONIUGATA = GILBERT. 14. SE GLI EPATOCITI SONO DANNEGGIATI, AUMENTANO SIA LA BILIRUBINA NON CONIUGATA CHE QUELLA CONIUGATA (EPATITE). 15. SE L’ESCREZIONE DELLA BILIRUBINA DAL POLO BILIARE DEGLI EPATOCITI E’ CONTRASTATA (FARMACI) O SE LE VIE BILIARI SONO “CHIUSE” (INFEZIONI, CALCOLI, TUMORI), AUMENTA LA BILIRUBINA CONIUGATA.
. 15. SE L’ESCREZIONE DELLA BILIRUBINA DAL POLO BILIARE DEGLI EPATOCITI E’ CONTRASTATA (FARMACI) O SE LE VIE BILIARI SONO CHIUSE (INFEZIONI, CALCOLI, TUMORI), AUMENTA LA BILIRUBINA CONIUGATA.")
58
EMOLISI PATOLOGICA RIDUZIONE DELLA VITA MEDIA ERITROCITARIA < 120 GIORNI COMPENSATA, SENZA ANEMIA: SE LA VITA MEDIA ERITROCITARIA E’ > 20 GIORNI SCOMPENSATA, CON ANEMIA: SE LA VITA MEDIA ERITROCITARIA E’ < 20 GIORNI O SE ERITROPOIESI RIDOTTA

59
EMOLISI PATOLOGICA - CLINICA
ITTERO URINE IPERCROMICHE FECI IPERCROMICHE SPLENOMEGALIA LITIASI BILIARE

63
EMOLISI PATOLOGICA - LABORATORIO
APTOGLOBINA BILIRUBINA NON CONIUGATA (INDIRETTA) LATTICO DEIDROGENASI (LDH) RETICOLOCITOSI (INDICE DI COMPENSO, NON DI EMOLISI)
 LATTICO DEIDROGENASI (LDH) RETICOLOCITOSI (INDICE DI COMPENSO, NON DI EMOLISI)")
64
3 categorie Hereditary Acquired

65
The morphology of RBC may provide evidence both of hemolysis and its cause

66
Classificazione anemie emolitiche
Anemie emolitiche congenite Da deficit enzimatici che causano una anomalia del metabolismo energetico Enzimi dello shunt degli esosomonofosfati e del metabolismo del glutatione Enzimi della via di Embden-Meyerhof Enzimi del metabolismo dei nucleotidi Emoglobinopatie Da alterazioni della membrana eritrocitaria Anemie emolitiche acquisite Su base immunitaria Su base non immunitaria

67
Classificazione anemie emolitiche
Enzimi dello shunt degli esosomonofosfati e del metabolismo del glutatione Glucosio -6-fosfato-deidrogenasi (G-6-PD) Glutatione perossidasi (GSH-Px) Deficit di Glutatione secondario a deficit di g-glutammil-cisteina sintetasi Glutatione-sintetasi Glutatione-reduttasi Enzimi della via di Embden-Meyerhof Esochinasi (HK) Glucosio-fosfato-isomerasi (GPI) Fosfofruttochinasi (PFK) Aldolasi (ALD) Trioso fosfato isomerasi (TPI) Difosfoglicerato mutasi (DPGM) Fosfoglicerato chinasi (PGK) Piruvato chinasi (PK) Enzimi del metabolismo dei nucleotidi Adenilato chinasi Pirimidina 5’-nucleotidasi
 Glutatione perossidasi (GSH-Px) Deficit di Glutatione secondario a deficit di g-glutammil-cisteina sintetasi. Glutatione-sintetasi. Glutatione-reduttasi. Enzimi della via di Embden-Meyerhof. Esochinasi (HK) Glucosio-fosfato-isomerasi (GPI) Fosfofruttochinasi (PFK) Aldolasi (ALD) Trioso fosfato isomerasi (TPI) Difosfoglicerato mutasi (DPGM) Fosfoglicerato chinasi (PGK) Piruvato chinasi (PK) Enzimi del metabolismo dei nucleotidi. Adenilato chinasi. Pirimidina 5’-nucleotidasi.")
68
Defects in Hexose-monophosphate shunt
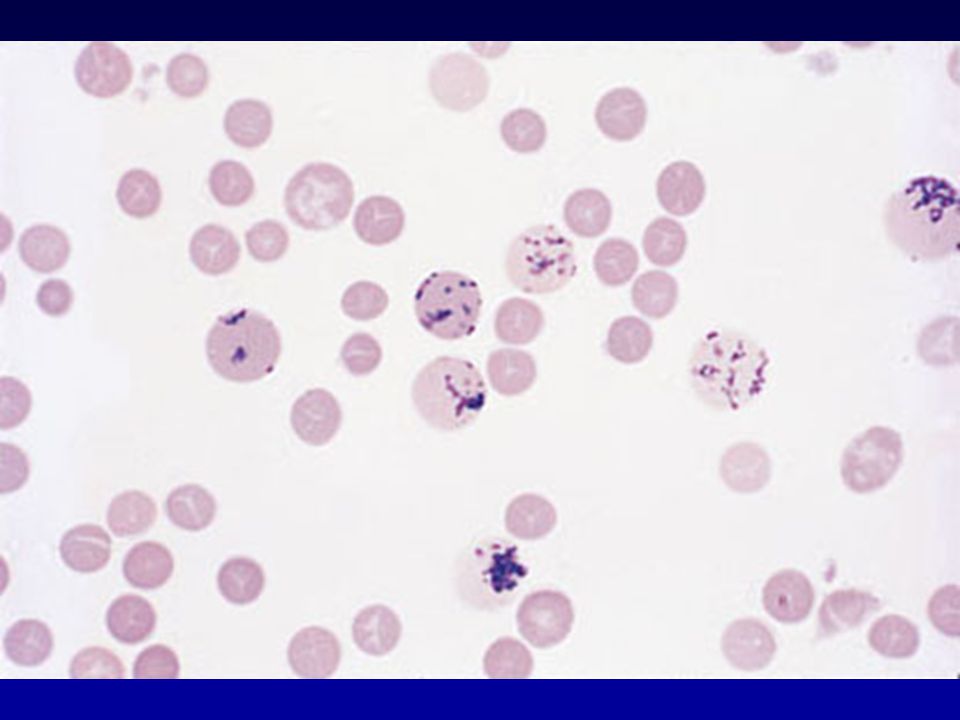
Via metabolica dedicata alla protezione del globulo rosso (Hb allo stato ridotto Fe++ e membrane) dagli agenti ossidanti Serve alla produzione di glutatione ridotto Difetti enzimatici impediscono mantenimento livelli adeguati di glutatione ridotto ----> Hb sulfidril groups oxidized ----> Heinz bodies
 dagli agenti ossidanti. Serve alla produzione di glutatione ridotto. Difetti enzimatici impediscono mantenimento livelli adeguati di glutatione ridotto ----> Hb sulfidril groups oxidized ----> Heinz bodies.")
69
Defects in Embden-Meyerhof pathway
Inherited in autosomal recessive pattern Low frequency RBC relatively deficient in ATP RBC are rigid, have K+ leaking channels, are sequestered by RE Deficiencies localized to RBC in most cases PK deficiency 95% cases Glucose phosphate isomerase 4%

71
METABOLIC DISEASES OF THE RBC: WHAT SHOULD BE KNOWN
Describe briefly the enzymatic pathways of RBC Know the basic functions of ATP, NADPH and 2,3PG in RBC Explain why many metabolic defects express mostly with anemia Describe epidemiology, pathogenesis and clinicakl features of GAPDH and PK deficiencies

Presentazioni simili













.>")












