Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
CLASSIFICAZIONE WHO DEI TUMORI DEL SISTEMA NERVOSO
T.neuroepiteliali T.dei nervi periferici T.delle meningi Linfomi e neoplasie ematopoietiche T.cellule germinali T.della sella T. metastatici

2
TUMORI DEL SNC Intracranici (intraparenchimali e extraparenchimali) Extracranici Intraparenchimali e extraparenchimali (primitivi e secondari) Citotipo Cellularità mista Differenziazione mista Sede (sopratentoriale,adulto; sottotentoriale,bambino)
")
3
TUMORI DEL SNC I tumori primitivi del SNC costituiscono il 9% di tutti i tumori Due picchi di eta’: prima decade e sesta-settima decade Classi sociali più elevate CLINICA (Sede, Crescita) Cefalea (aumento pressione intracranica) Epilessia (tumori infiltranti) Falsi segni focali (paralisi dei nervi III e VI) per erniazioni e pressione Segni focali e insufficienze funzionali a lenta progressione Edema, emorragia, idrocefalo
 Cefalea (aumento pressione intracranica) Epilessia (tumori infiltranti) Falsi segni focali (paralisi dei nervi III e VI) per erniazioni e pressione. Segni focali e insufficienze funzionali a lenta progressione. Edema, emorragia, idrocefalo.")
4
Caratteristiche biologiche
Malignità biologica /malignità istologica Infiltranti Disseminazione intraneurale Metastasi extraneurali (non comuni) Difficoltà diagnostiche Tumori con caratteristiche istologiche simili Diversa origine Diversa prognosi
 Difficoltà diagnostiche. Tumori con caratteristiche istologiche simili. Diversa origine. Diversa prognosi.")
5
Classificazione WHO 2007

6
TUMORI NEUROEPITELIALI
Tumori astrocitici Tumori oligodendrogliali Tumori oligoastrocitari Tumori ependimali Tumori del plesso coroideo Altri tumori neuroepiteliali Tumori neuronali e misti neuronali-gliali Tumori del parenchima pineale Tumori embrionali (MB, CNS PNET, ATRT)
")
7
Tumori astrocitici Astrocitoma diffuso Fibrillare Gemistocitico Protoplasmatico Astrocitoma anaplastico Glioblastoma Astrocitoma pilocitico Xantoastrocitoma pleomorfo Astrocitoma subependimale a c.giganti Gliomatosis cerebri

8
Astrocitomi diffusamente infiltranti
Emisferici Diffusamente infiltranti Adulto Variabilità istopatologica 60% di tutti i t.primitivi del SNC

9
Caratteristiche istopatologiche: grading (St.Anne/Mayo)
WHO StAnne/Mayo I Astro pilocitico II Astro diffuso Astro Grado 2 Atipia nucleare III Astro anapl Astro Grado 3 + mitosi IV glioblastoma Astro Grado 4 + prol.endot + necrosi

10
Sopravvivenza Età Sede e trattamento (radioterapia, resezione chirurgica, chemio) Eziologia Radiazioni Sequenze DNA SV40 large T-antigen Molecolare (accumulo di lesioni genetiche riguardanti la proliferazione cell; oncogeni/oncosoppressori) Grado II a III: perdita di allele 9p,13q,amplificazione12q (9p21 geni codificanti per p16 e p15 del ciclo cell)
 Grado II a III: perdita di allele 9p,13q,amplificazione12q (9p21 geni codificanti per p16 e p15 del ciclo cell)")
11
ASTROCITOMA DIFFUSO Elevata differenziazione cellulare, lenta crescita; Età: ; Sesso: maschi > femmine Grado: II Localizzazione: supratentoriale, frontale e temporale Macro: margini indistinti o massa a confini indefinibili Istopat: cellularità, rare atipie nucl., mitosi assenti,GFAP+ fibrillari, gemistocitici (citoplas.eosinofilo e fini processi), protoplasmatici (piccolo corpo cell., scarsi e flaccidi processi, deg.mucoide
, protoplasmatici (piccolo corpo cell., scarsi e flaccidi processi, deg.mucoide.")
13
ASTROCITOMA

16
Sopravvivenza: 6-8 anni Fattori prognostici: età, resezione chirurgica, mutazione TP53 – (variante gemistocitica)
")
17
ASTROCITOMA ANAPLASTICO
Anaplasia focale o diffusa; marcato potenziale proliferativo; progressione in glioblastoma Età: media 41 Sesso: maschi > femmine Grado: III Localizzazione: emisfero cerebrale Macro: indistinguibile dall’astro diffuso Istopat: cellularità + elevata, atipie nucl., mitosi frequenti

18
Genetica molecolare: mutazione TP53
Sopravvivenza: 3 anni Fattori prognostici: età, resezione chirurgica, componente oligo +

21
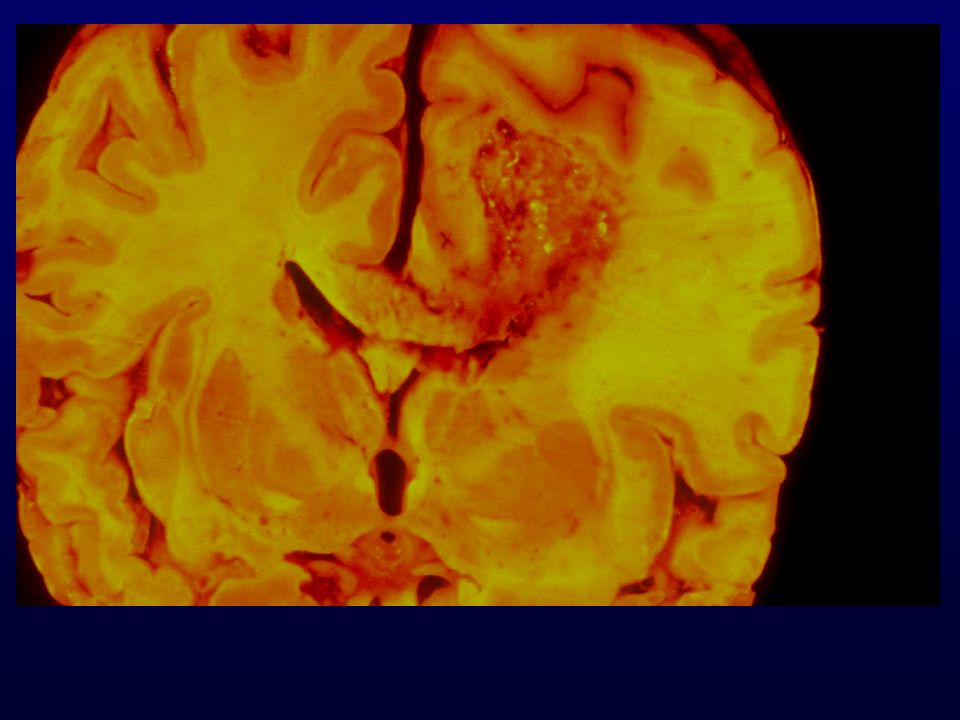
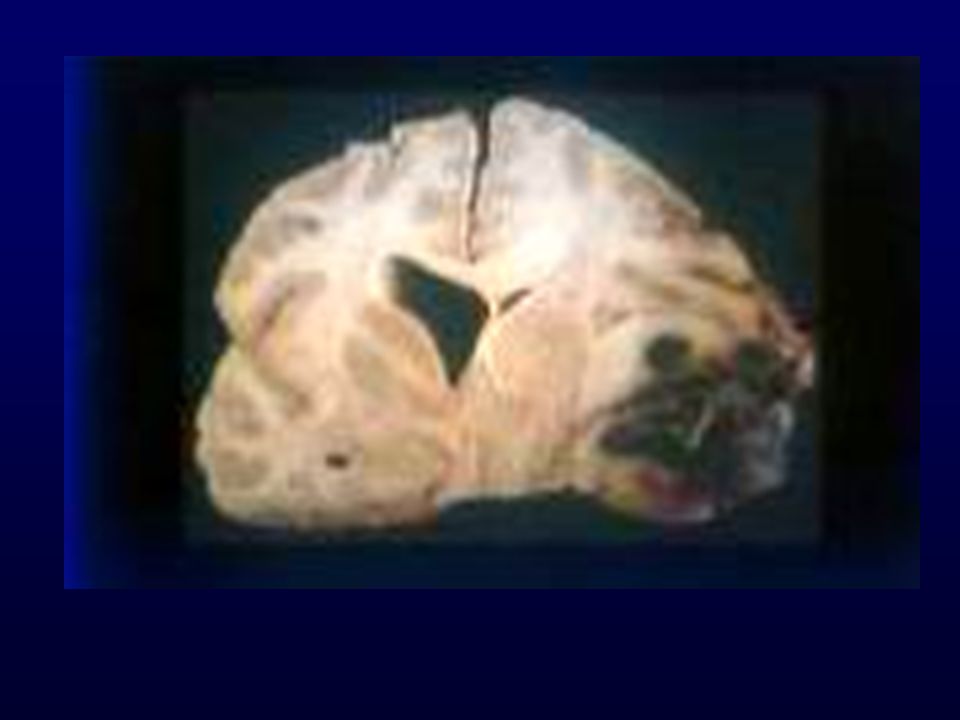
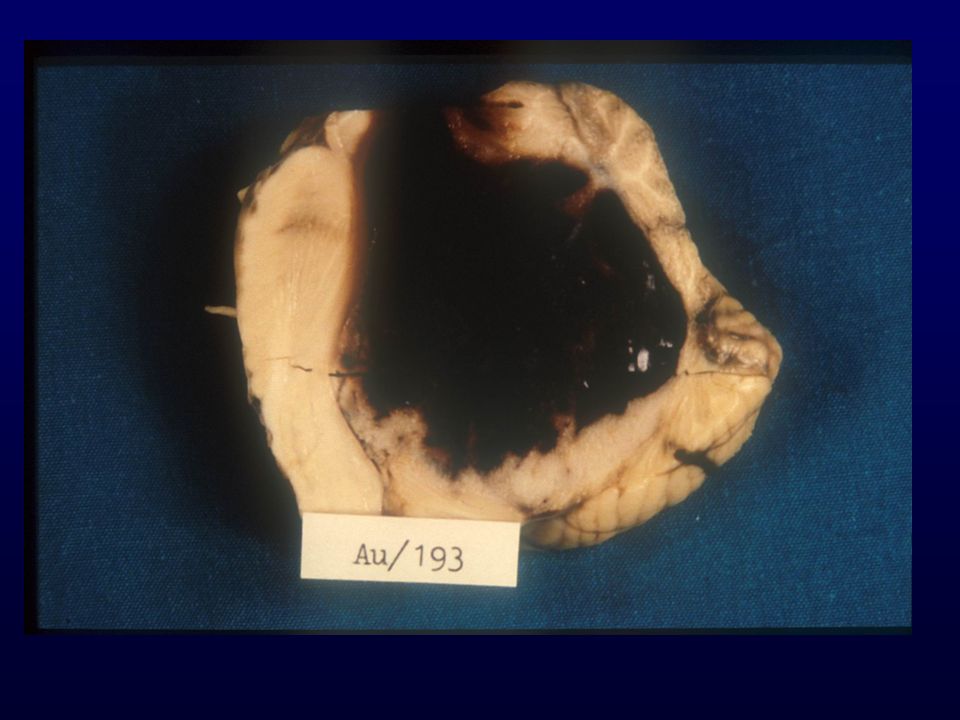
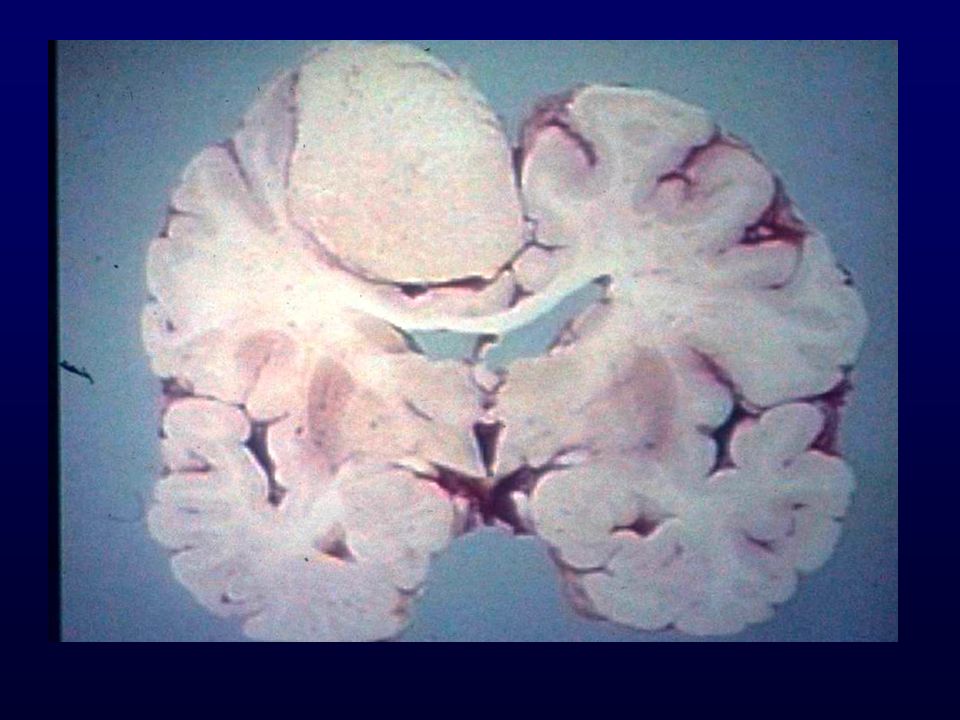
GLIOBLASTOMA Astrociti scarsamente diff., proliferazione vascolare, necrosi; progressione da grado III o insorgenza “de novo”; Età: media (rare manifestazioni in utero); Incidenza: 12-15% SNC Sesso: maschi > femmine Grado: IV Localizzazione: sostanza bianca subcorticale dell’emisfero cerebrale (temporale, parietale, frontale) Macro: massa grigiastra mal definita, necrosi, emorragie, unilaterale o bilaterale (a farfalla) Diffusione: rapida, attraverso il corpo calloso, rare metastasi via liquor, rare metastasi via ematica (iatrogena)
; Incidenza: 12-15% SNC. Sesso: maschi > femmine. Grado: IV. Localizzazione: sostanza bianca subcorticale dell’emisfero cerebrale (temporale, parietale, frontale) Macro: massa grigiastra mal definita, necrosi, emorragie, unilaterale o bilaterale (a farfalla) Diffusione: rapida, attraverso il corpo calloso, rare metastasi via liquor, rare metastasi via ematica (iatrogena)")
23
GLIOBLASTOMA – INFILTRAZIONE EMISFERO CONTROLATERALE

29
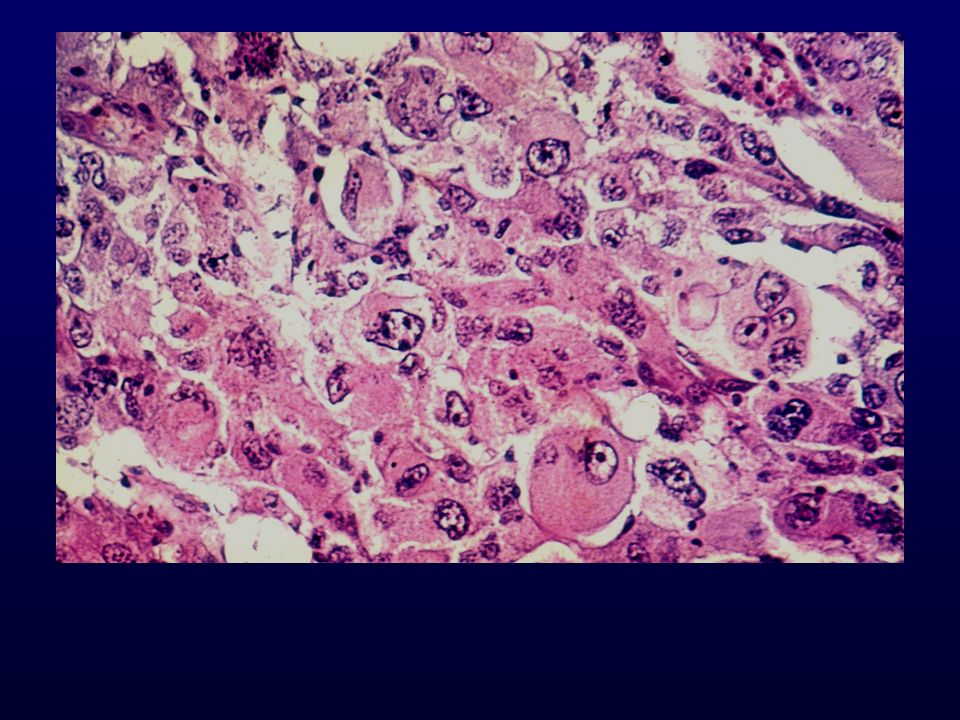
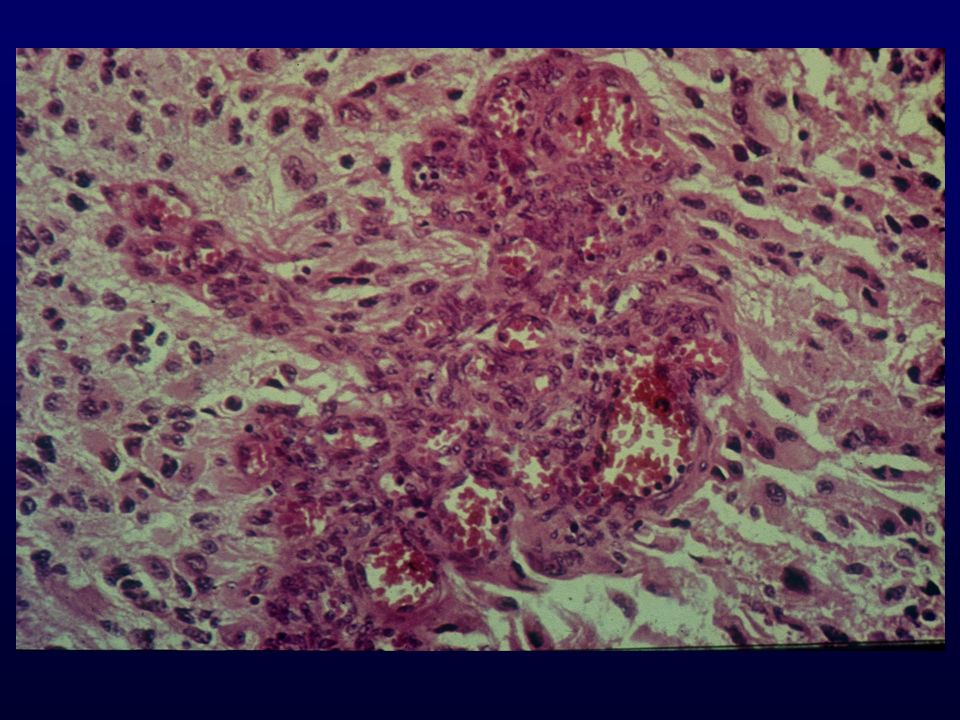
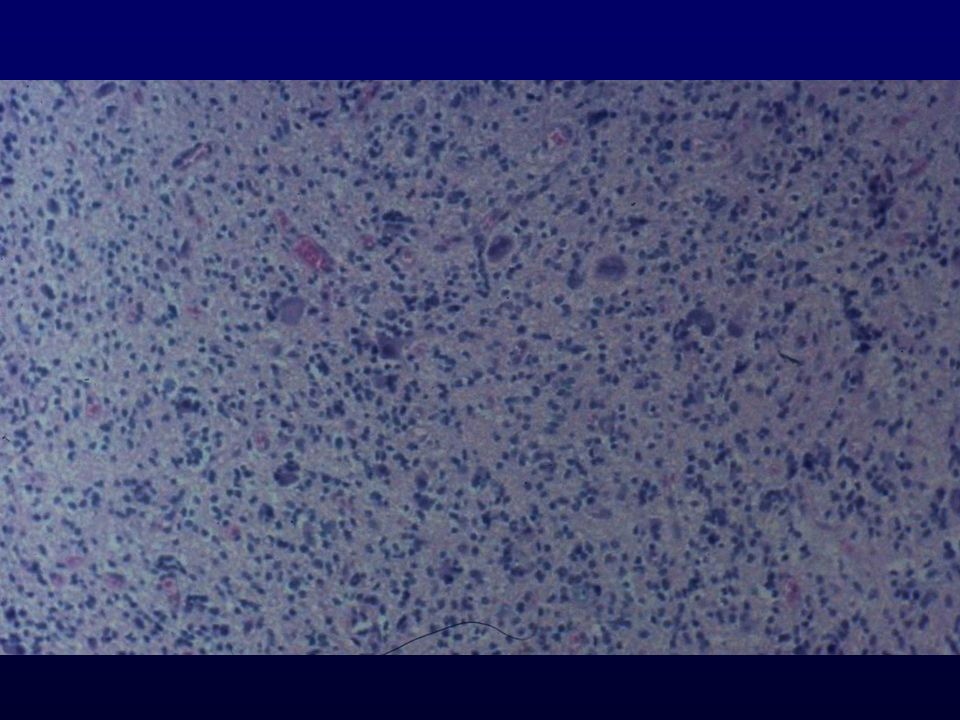
Istopatologia: astrociti poco diff
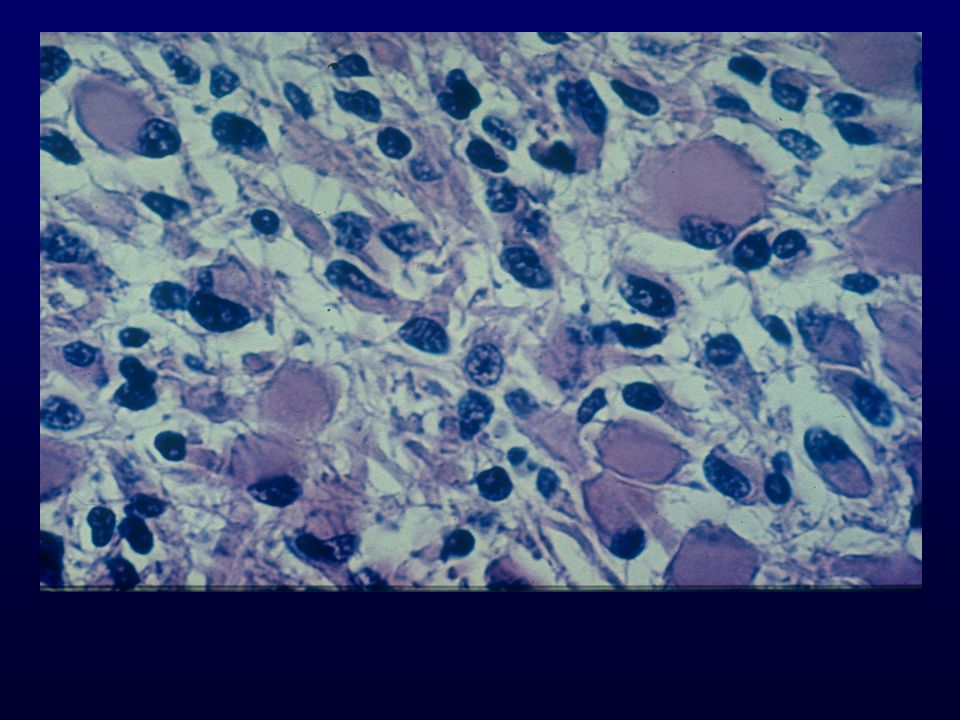
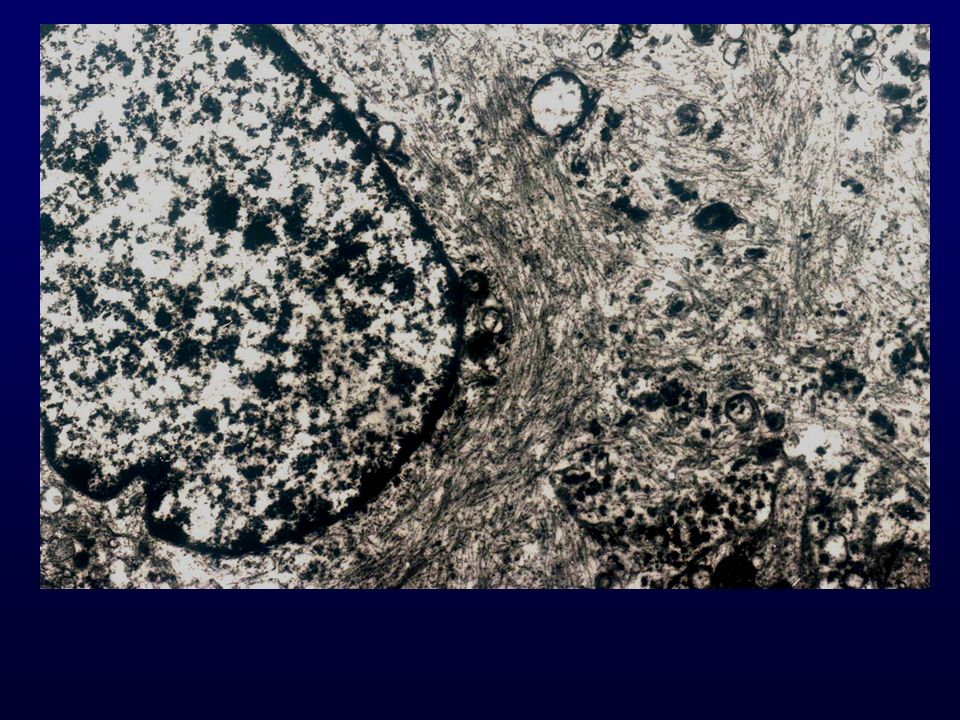
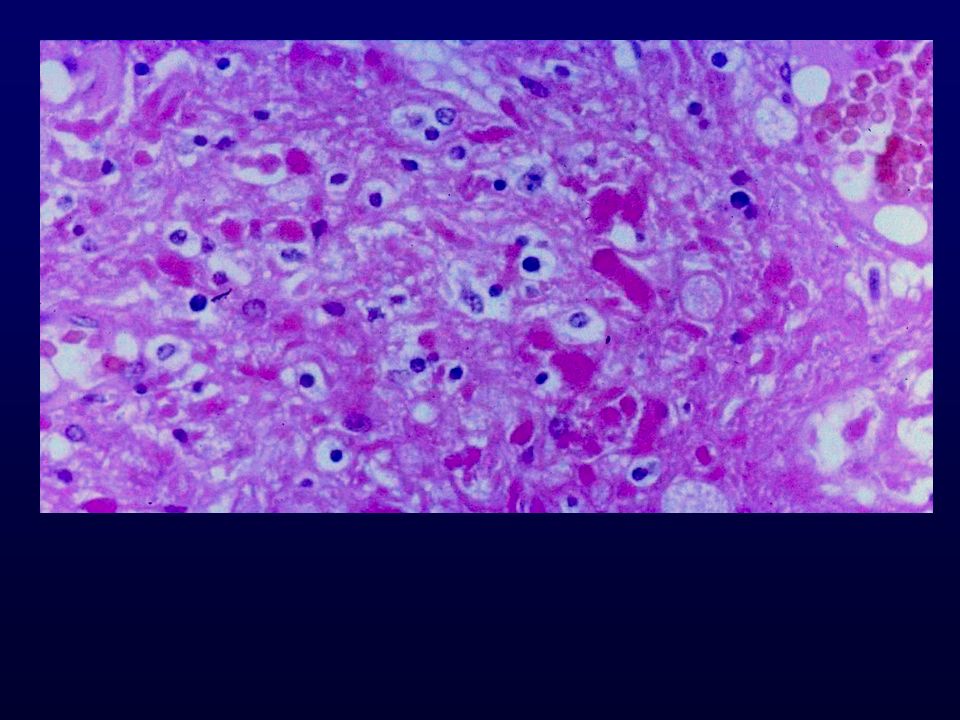
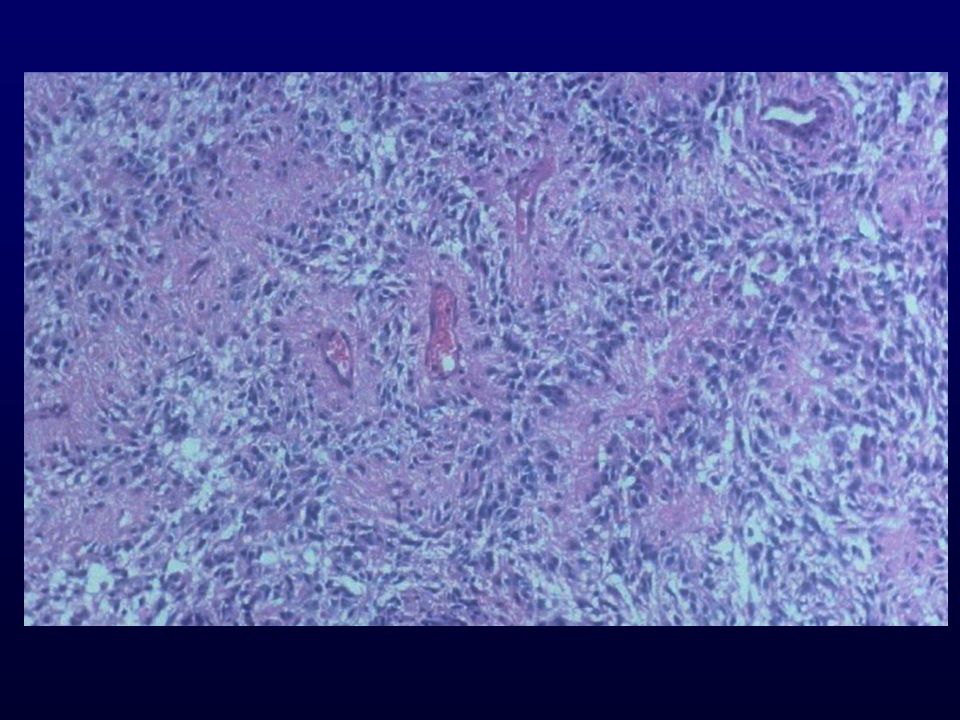
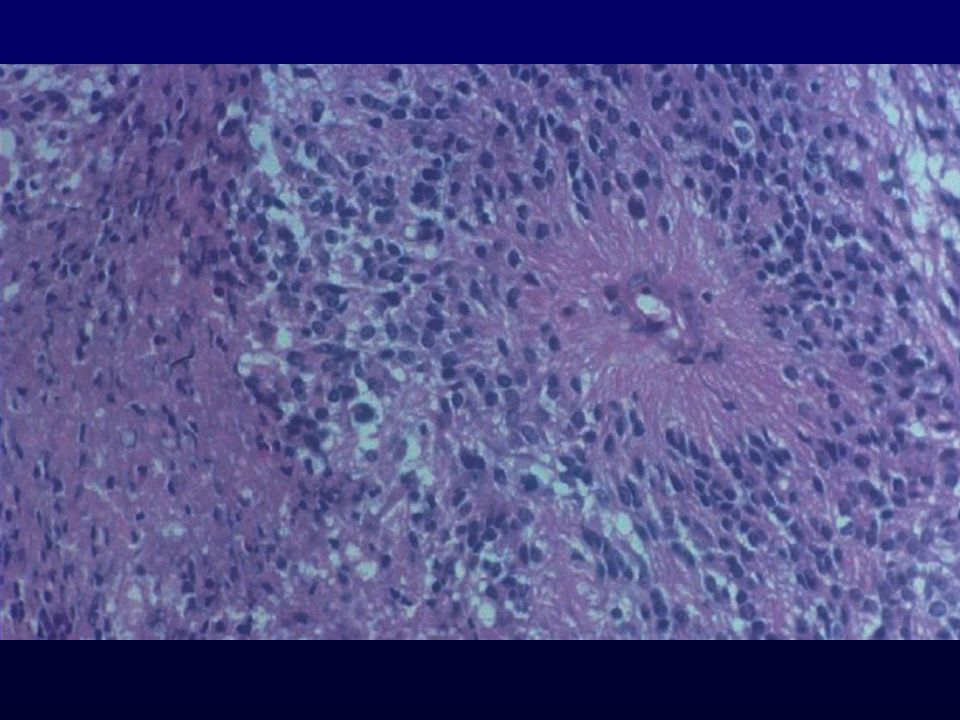
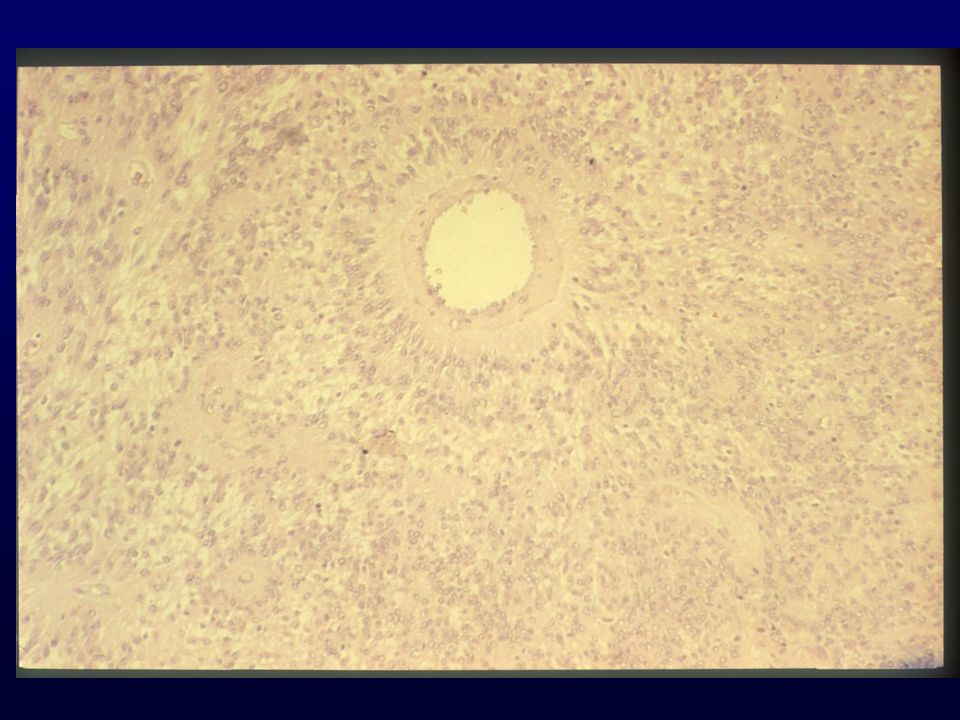
Istopatologia: astrociti poco diff., atipia nucleare, marcata attività mitotica, proliferazione microvascolare (aspetto glomeruloide), necrosi (a palizzata), strutture ghiandolari, cell.giganti multinucleate, cell.lipidizzate, cell.granulari, GFAP+ EM: variabilità morfologica, mitocondri degenerati, presenza filamenti dipende da differenziazione Genetica molecolare: mutazione TP53, LOH 17p,(progressione da grado III); LOH 10p, amplificazione EGFR (GBM de novo) Sopravvivenza: <1 anno indipendente da chemio- e radioterapia Fattori prognostici: età (< 45 anni), necrosi -, resezione chirurgica, EGFR -, PTEN+, TP53 indiff.
, necrosi (a palizzata), strutture ghiandolari, cell.giganti multinucleate, cell.lipidizzate, cell.granulari, GFAP+ EM: variabilità morfologica, mitocondri degenerati, presenza filamenti dipende da differenziazione. Genetica molecolare: mutazione TP53, LOH 17p,(progressione da grado III); LOH 10p, amplificazione EGFR (GBM de novo) Sopravvivenza: <1 anno indipendente da chemio- e radioterapia. Fattori prognostici: età (< 45 anni), necrosi -, resezione chirurgica, EGFR -, PTEN+, TP53 indiff.")
30
ASTROCITOMA PILOCITICO
Circoscritto, lenta crescita, pattern bifasico (cell.bipolari + fibre di Rosenthal; cell.multipolari + microcisti e granular bodies) Età: anni Clinica: cefalea, endocrinopatie, deficit visivi, no epilessia Sesso: no prevalenza Grado: I Localizzazione: nevrasse; nervo ottico, chiasma ottico, cervelletto,emisfero cerebrale, midollo spinale Macro: massa discreta con cisti Istopat: pattern bifasico, scarsa cellularità, processi hair-like, rare mitosi, prol.vasc, alterazioni regressive (calcificazioni, necrosi, linfociti), GFAP +, fibre di Rosenthal (αβcristallina), EGB (α1chimotripsina e α1tripsina)
 Età: anni. Clinica: cefalea, endocrinopatie, deficit visivi, no epilessia. Sesso: no prevalenza. Grado: I. Localizzazione: nevrasse; nervo ottico, chiasma ottico, cervelletto,emisfero cerebrale, midollo spinale. Macro: massa discreta con cisti. Istopat: pattern bifasico, scarsa cellularità, processi hair-like, rare mitosi, prol.vasc, alterazioni regressive (calcificazioni, necrosi, linfociti), GFAP +, fibre di Rosenthal (αβcristallina), EGB (α1chimotripsina e α1tripsina)")
35
Genetica: correlato a NF1
Sopravvivenza: benigno; a lenta evoluzione con possibilità di stabilizzazione o regressione, associazione a NF1 più aggressivo; associato a rare mitosi, aumentata cellularità, atipia cellulare (astro pilocitico atipico) non ha significato clinico
 non ha significato clinico.")
36
Tumori oligodendrogliali
Oligodendroglioma Oligodendroglioma anaplastico Gliomi misti Oligoastrocitoma Oligoastrocitoma anaplastico

37
OLIGODENDROGLIOMA Cellule oligodendrogliali ben diff; diffusamente infiltrante; Età: anni, rari nell’infanzia; Incidenza: 4.2% SNC Sesso: maschi > femmine Grado: II Localizzazione: sostanza bianca emisferica (frontale) Clinica: convulsioni epilettiche, cefalea Macro: massa soffice, grigio-rosata o gelatinosa, calcificazioni Istopat: moderatamente cellulato, cell.rotonde con citoplasma chiaro, microcalcificazioni, rete capillare, occasionali mitosi; no markers specifici (S-100, Leu7, GFAP, Vim EM: cellule rotonde, citoplasma estratto, nucleo regolare, microtubuli perinucleari, tozzi e corti microvilli, aggregati mitoc
 Clinica: convulsioni epilettiche, cefalea. Macro: massa soffice, grigio-rosata o gelatinosa, calcificazioni. Istopat: moderatamente cellulato, cell.rotonde con citoplasma chiaro, microcalcificazioni, rete capillare, occasionali mitosi; no markers specifici (S-100, Leu7, GFAP, Vim. EM: cellule rotonde, citoplasma estratto, nucleo regolare, microtubuli perinucleari, tozzi e corti microvilli, aggregati mitoc.")
40
Genetica molecolare: LOH 19q, 1p; forte espressione di EGFR
Sopravvivenza: 3-5 anni Fattori prognostici: età, localizzazione frontale, resezione chirurgica

41
Tumori ependimali Ependimoma Cellulare Papillare A cellule chiare Tanicitico Ependimoma anaplastico Ependimoma mixopapillare Subependimoma

42
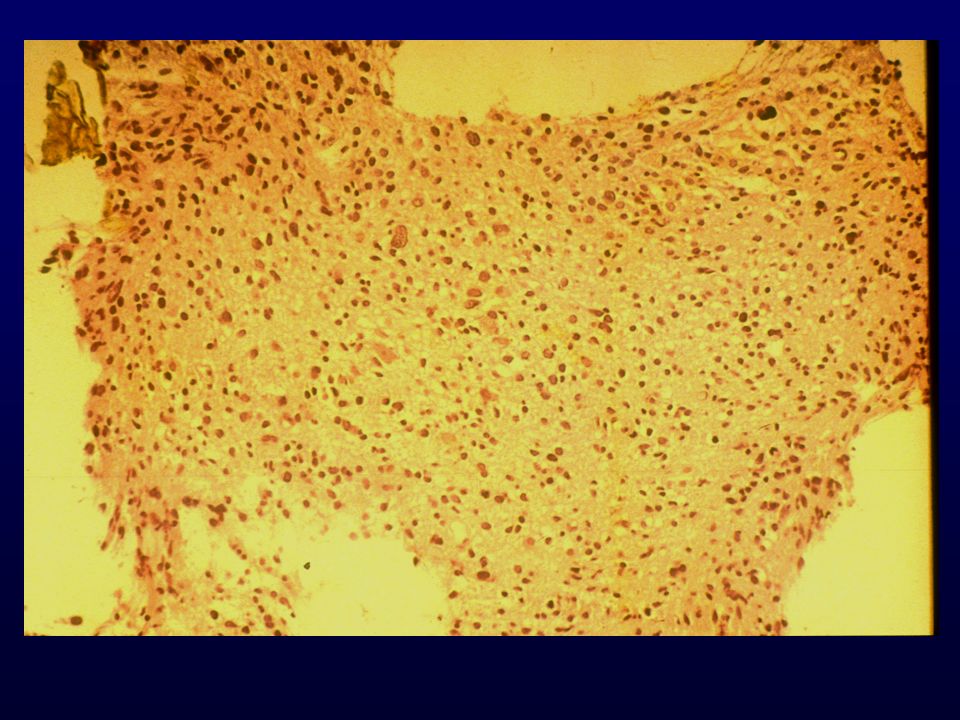
EPENDIMOMA Cellule ependimali ben diff; lenta crescita; bambino e giovane adulto; parete dei ventricoli; Incidenza: 3-9% dei t. neuroepiteliali Età: bambino, infratentoriale 30-40 anni, spinale Bambino e adulto, sopratentoriale Sesso: non prevalenza Grado: II Localizzazione: fossa posteriore e midollo spinale; III,IV e ventricoli laterali Clinica: dipende dalla sede (sottotentoriali, idrocefalo; fossa post, atassia cerebellare; sopratentoriali, deficit neurologici, convulsioni)
")
44
Macro: ben demarcato, soffice, grigiastro-rosa, cisti, foci di necrosi o emorragici
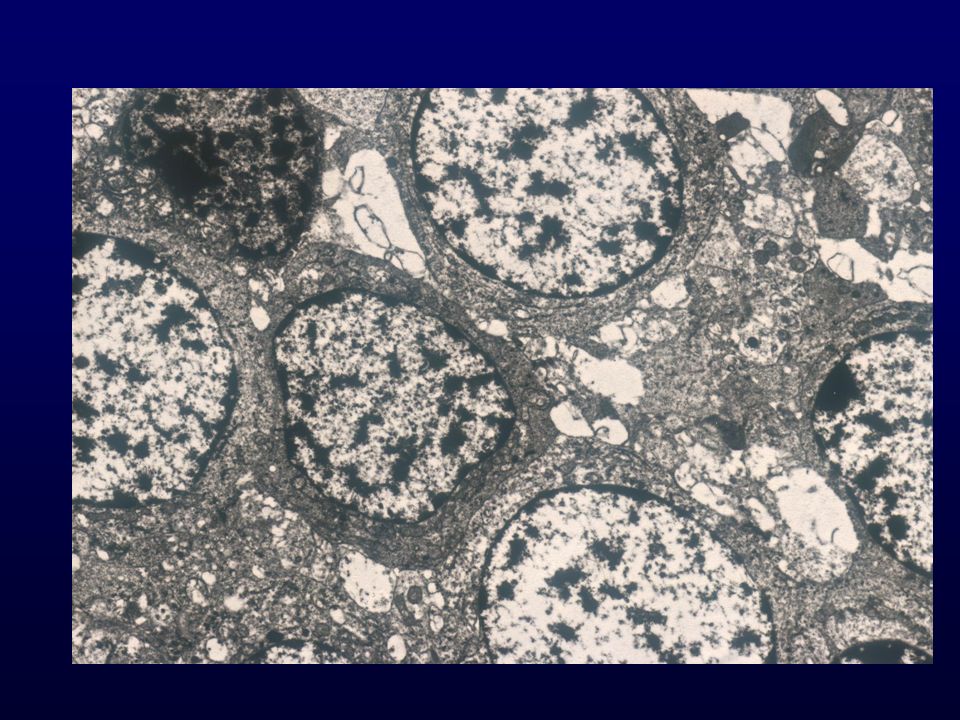
Istopatologia: nuclei monomorfi, pseudorosette perivascolari e rosette ependimali, mitosi rare o assenti, necrosi occasionale; GFAP, Vim., S-100, EMA, CK focali Cellulare, scarse rosette; Papillare, papille ben formate Cellule chiare, DD. oligo, neurocitoma; EM diagnostica Tanicitico, aspetto astrocitico EM: cellule fusate, filamenti gliali, giunzioni zipper-like, microrosette con microvilli, ciglia

49
Genetica: correlato a NF2
Genetica molecolare: LOH 17p Sopravvivenza: 5-10 anni indipendente dal grado di resezione chirurgica Fattori prognostici: giovane età -, localizzazione (spinale, sovratentoriale, fossa)
")
50
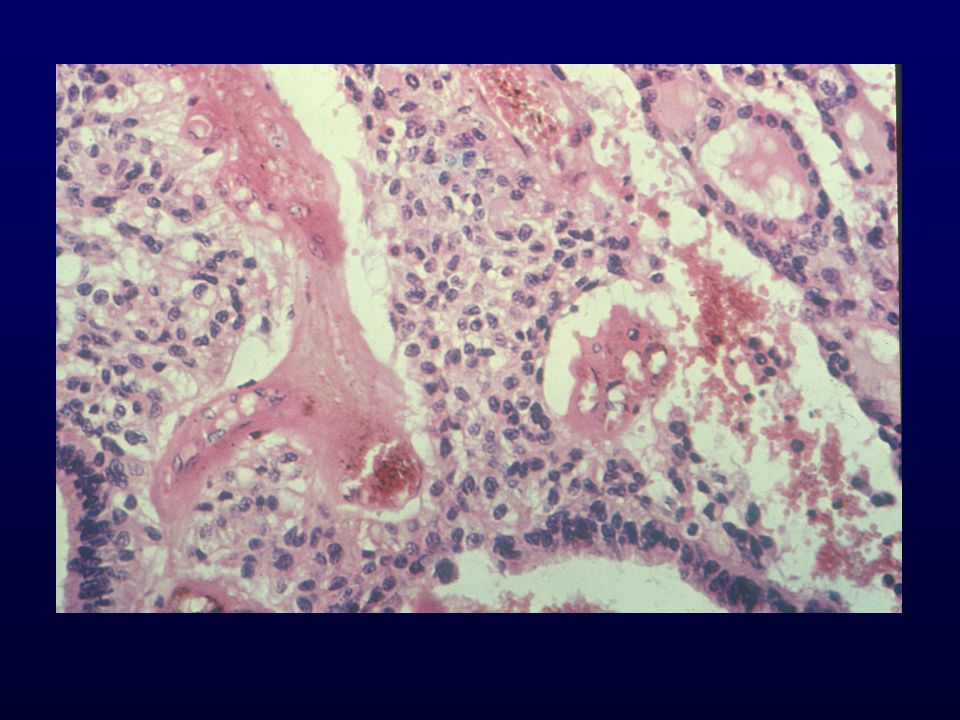
EPENDIMOMA MIXOPAPILLARE
Giovane adulto, filum terminale della cauda, pattern papillare Incidenza: 13% degli ependimomi; neoplasia intramidollare + comune Età: media 36 Sesso: femmine>maschio Grado: I Localizzazione: cauda, raramente emisferico Clinica: dolori alla schiena di lunga durata Macro: soffice, lobulato, grigiastro Istopatologia: pattern papillare con cell.cuboidali, matrice mixoide, mitosi rare o assenti; ICC GFAP, S-100, Vim EM: caratteristiche ependimali, lamina basale

52
Sopravvivenza: prognosi buona, 10 anni indipendente dal grado di resezione chirurgica; recidive rare

53
PAPILLOMA DEI PLESSI Eta pediatrica Asse connettivale ricoperto da epitelio cuboidale a volte colonnare Idrocefalo (anche comunicante per iperproduzione)
")
56
Tumori neuronali e misti neuronali-gliali
Gangliocitoma Astrocitoma infantile desmoplastico/ Ganglioglioma DNT, tumore neuroepiteliale disembrioplastico Ganglioglioma Ganglioglioma anaplastico Neurocitoma centrale Liponeurocitoma cerebellare Paraganglioma

57
NEUROCITOMA CENTRALE Cellule rotonde a diff neuronale; ventricolo laterale;giovane adulto; prognosi favorevole Incidenza: % SNC Età: media 29 anni Sesso: non prevalenza Grado: II Localizzazione: ventricoli laterali e III ventricolo Clinica: pressione intracranica, occasionali deficit visivi e mentali Macro: massa grigiastra, friabile, calcificazioni e occasionali emorragie

59
Istopatologia: cellule rotonde, monomorfe, oligo-like; neuropilo (aree fibrillari); microvascolarizzazione con aspetti di arborizzazione; ICC : Sinaptofisina (falsi + o negatività) DD oligodendroglioma, ependimoma a cell.chiare, pineocitoma, DNT EM: cellule rotonde, nucleo monomorfo, microtubuli dispersi, granuli neuroendocrini, prolungamenti neuritici con microtubuli, strutture simil-sinaptiche Genetica molecolare: gain chr.7, isocromosoma 17 Sopravvivenza: prognosi favorevole, resezione completa; può avere aggressività istologica non influente sulla prognosi

60
GANGLIOGLIOMA Gruppi irregolari di neuroni displastici, multipolari + componente gliale costituita da astrociti circondati da stroma reticolinico. Necrosi assente Incidenza: 0.4% SNC Età: media tra 8.5 e 25 anni Sesso: 1.1:1 Grado: I Localizzazione: cervello, cervelletto, midollo allungato, spinale, nervo ottico, ipofisi, gh. pineale Clinica: dipende dalla sede Macro: massa solida o cistica, occasionali calcificazioni

63
T.rabdoide-teratoide atipico
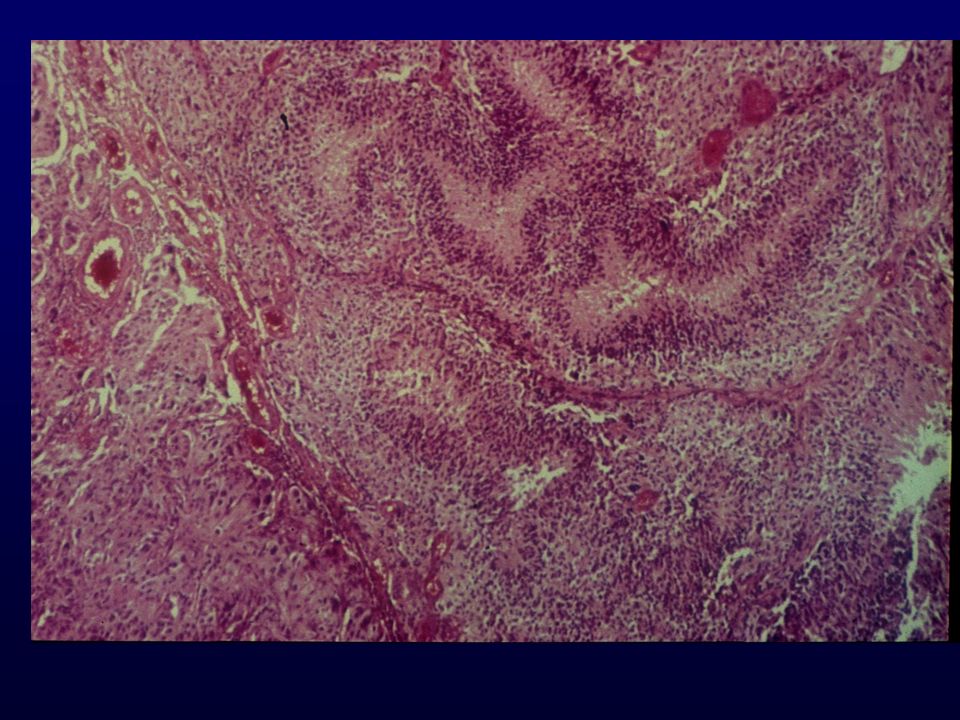
TUMORI EMBRIONALI Medulloepitelioma Ependimoblastoma Medulloblastoma Medulloblastoma desmoplastico Medulloblastoma a grandi cellule Medullomioblastoma Medulloblastoma melanotico PNET Neuroblastoma Ganglioneuroblastoma T.rabdoide-teratoide atipico

64
MEDULLOBLASTOMA Tumore invasivo, maligno embrionale del cervelletto; differenziazione prevalentemente neuronale; bambino; tendenza alla disseminazione mediante liquor Incidenza: 0.5 x Età: media 7 anni; nell’adulto, anni Sesso: 65% nei maschi Grado: IV Localizzazione: verme del cervelletto; tendenza ad infiltrare il IV ventricolo Clinica: atassia del tronco, disturbo del passo, ipertensione secondaria ad ostruzione del canale ependimale, cefalea, vomito mattutino

66
Macro: consistenza variabile, emorragia massiva
Istopatologia: Medulloblastoma classico, cell.rotondo-ovali. Nucleo ipercromatico, scarso citoplasma, rosette neuroblastiche Medulloblastoma desmoplastico,noduli a bassa cell.con nucleo monomorfo, circondati da aree intensamente cellulate e proliferanti Medulloblastoma a grandi cellule, cell.grandi, nucleo pleomorfo, nucleoli prominenti, necrosi, elevata attività mitotica ICC: nestina, Vim, Sin, NF, GFAP, N-CAM EM: differenziazione neuroblastica con neuriti, granuli neuroendocrini, strutture simil-sinaptiche; aree di differenziazione gliale Genetica: associato a forme famigliari, sindrome di Turcot

69
Genetica molecolare: amplificazione gene MYC; LOH 17p, 1q, 10q,5q (gene APC)
Sopravvivenza: 50-70% a 5 anni Fattori negativi: età<3 anni, metastasi alla presentazione, resezione chirurgica parziale, variante a grandi cellule, differenziazione gliale, LOH 17p, amplificazione MYC Fattori positivi: variante desmoplastica, aneuploidia, differenziazione neuronale, apoptosi

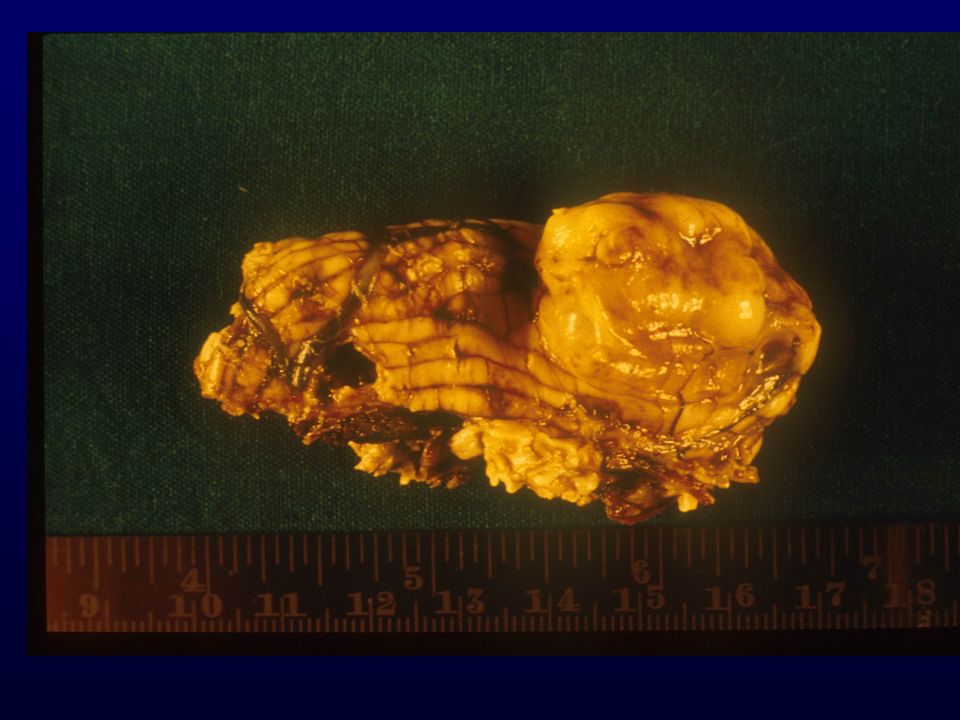
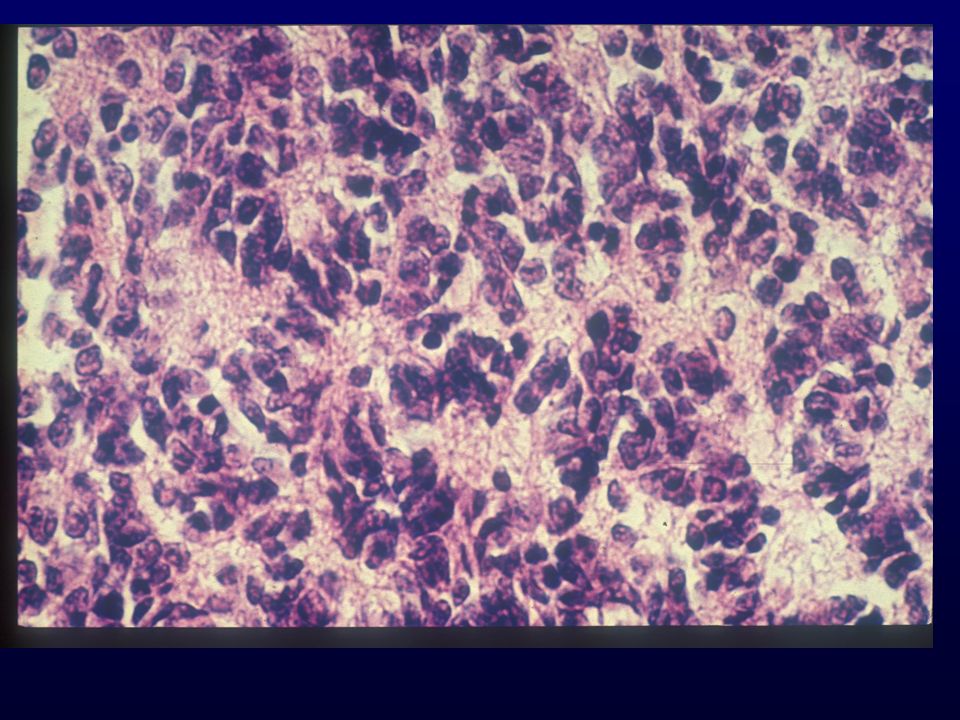
70
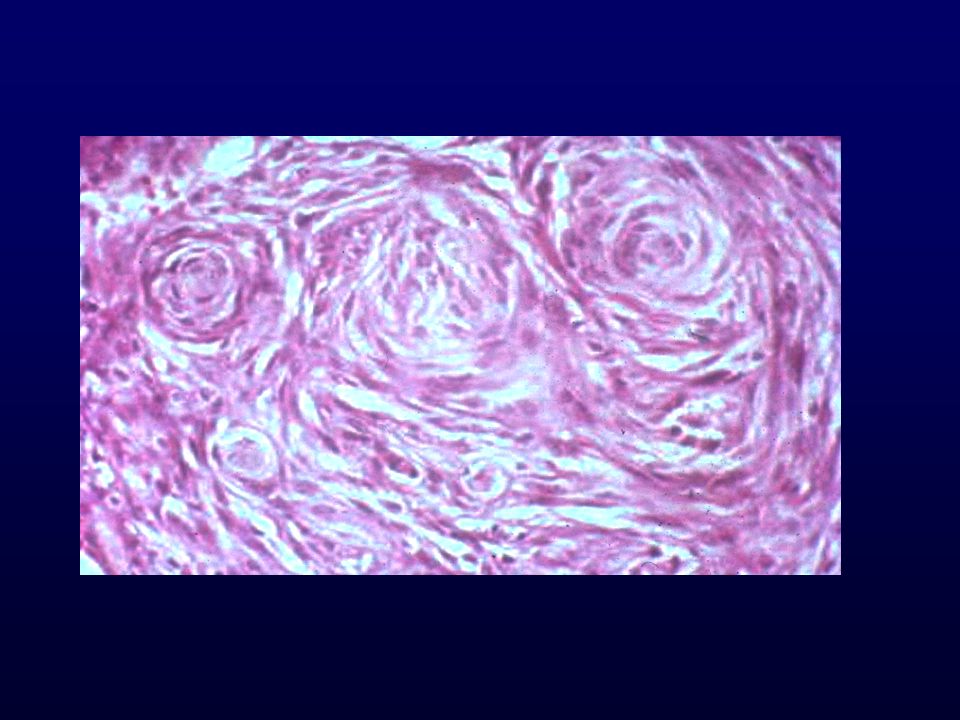
MENINGIOMA Tumore generalmente a lenta crescita, benigno in rapporto alla dura madre costituito da cellule meningoteliali (aracnoidali) . Può essere multifocale e recidivare Incidenza: 6 x (frequenza all’autopsia 1.4%) Età: adulto-anziano Sesso: femminile (rapporto 2:1) Grado: I, II (meningioma atipico), III (meningioma anaplastico) Localizzazione: intracranici (sopra le convessità cerebrali attaccati alla falce), cavità orbitale e intravertebrale, rari extracranici Clinica: sintomatologia da compressione delle strutture adiacenti o deficits specifici relativamente alla sede
 . Può essere multifocale e recidivare. Incidenza: 6 x (frequenza all’autopsia 1.4%) Età: adulto-anziano. Sesso: femminile (rapporto 2:1) Grado: I, II (meningioma atipico), III (meningioma anaplastico) Localizzazione: intracranici (sopra le convessità cerebrali attaccati alla falce), cavità orbitale e intravertebrale, rari extracranici. Clinica: sintomatologia da compressione delle strutture adiacenti o deficits specifici relativamente alla sede.")
71
Macro: consistenza solida o elastica; lobulato
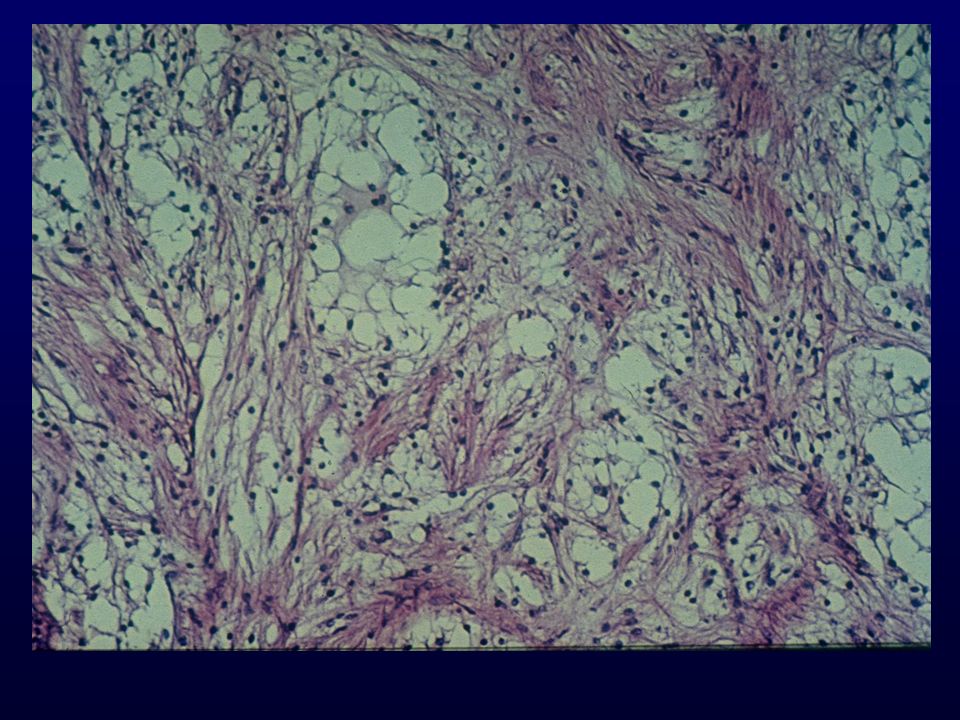
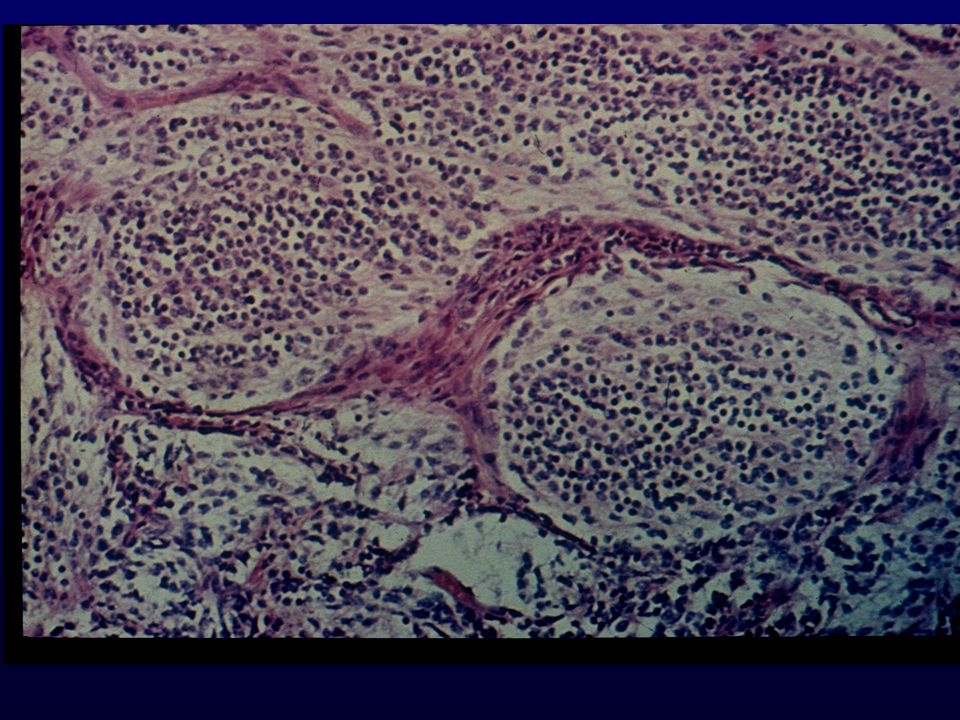
Istopatologia: Meningoteliale o sinciziale, whorls e corpi psammomatosi Fibroso, cellule fusate in fasci separati da abbondante collagene Transizionale, intermedio tra meningoteliale e fibroso Meningioma atipico, mitosi + 3 o piu parametri (cellularita, elevato N/C, nucleoli prominenti, necrosi, sheet-like), Meningioma anaplastico, simil-sarcoma, >20 mitosi x 10HPF Varianti: Psammomatosa, angiomatoso, microcistico,secretorio, metaplastico ,cordoide (II), a cellule chiare (II) , papillare (III), rabdoide (III)
, Meningioma anaplastico, simil-sarcoma, >20 mitosi x 10HPF. Varianti: Psammomatosa, angiomatoso, microcistico,secretorio, metaplastico ,cordoide (II), a cellule chiare (II) , papillare (III), rabdoide (III)")
72
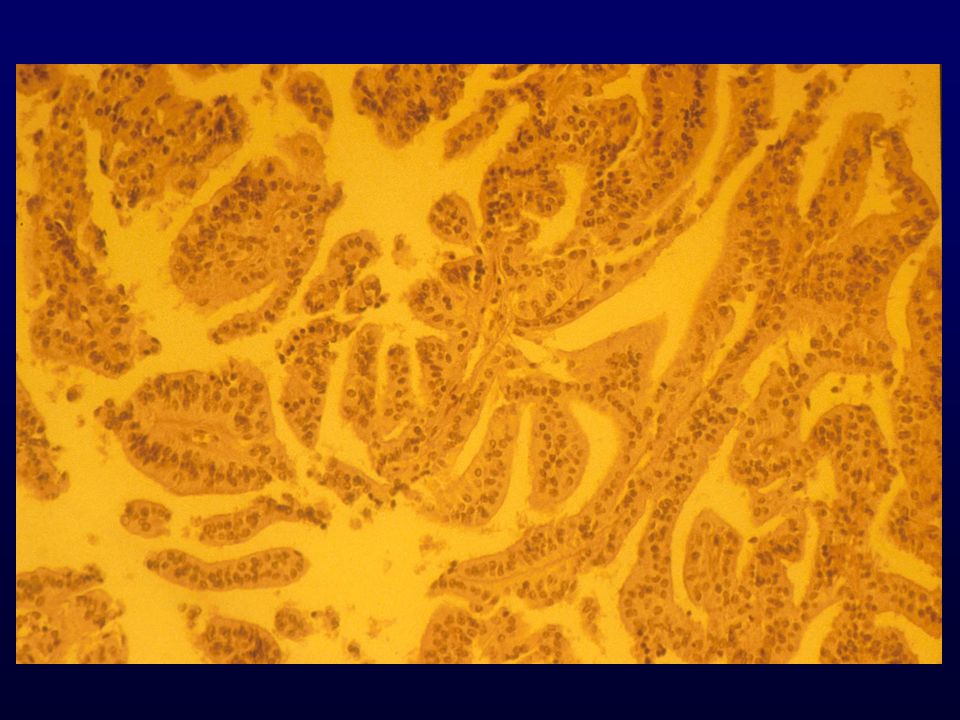
ICC: EMA, VIM, S-100, CEA (secretorio)
EM: desmosomi, prolungamenti citoplasmatici interdigitati, filamenti intermedi, nuclei con pseudoinclusioni. Genetica: Meningioma, grado I – LOH 22q, mutazioni NF2 Meningioma atipico, grado II – LOH 1p, 6q, 10q, 18q; gain 1q, 9q, 12q, 15q, 17q, 20q. Meningioma anaplastico, grado III – LOH 6q, 9p, 10 e 14 q, amplificazione 17q, mutazioni rare TP53, PTEN, delezioni rare CDKN2A PROGNOSI: predizione di recidiva e di sopravvivenza (grado III); estensione della resezione, sede, età, grado, indici di proliferazione (MIB1), recettori per il progesterone.
; estensione della resezione, sede, età, grado, indici di proliferazione (MIB1), recettori per il progesterone.")
74
CORPI PSAMMOMATOSI

77
MENINGIOMA A CELLULE CHIARE

78
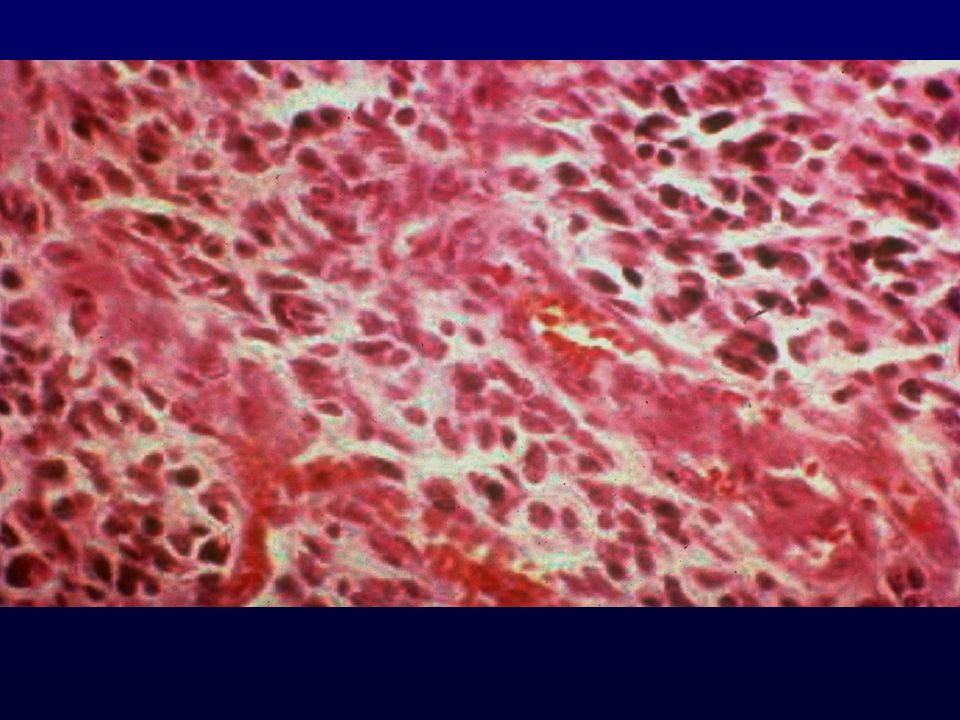
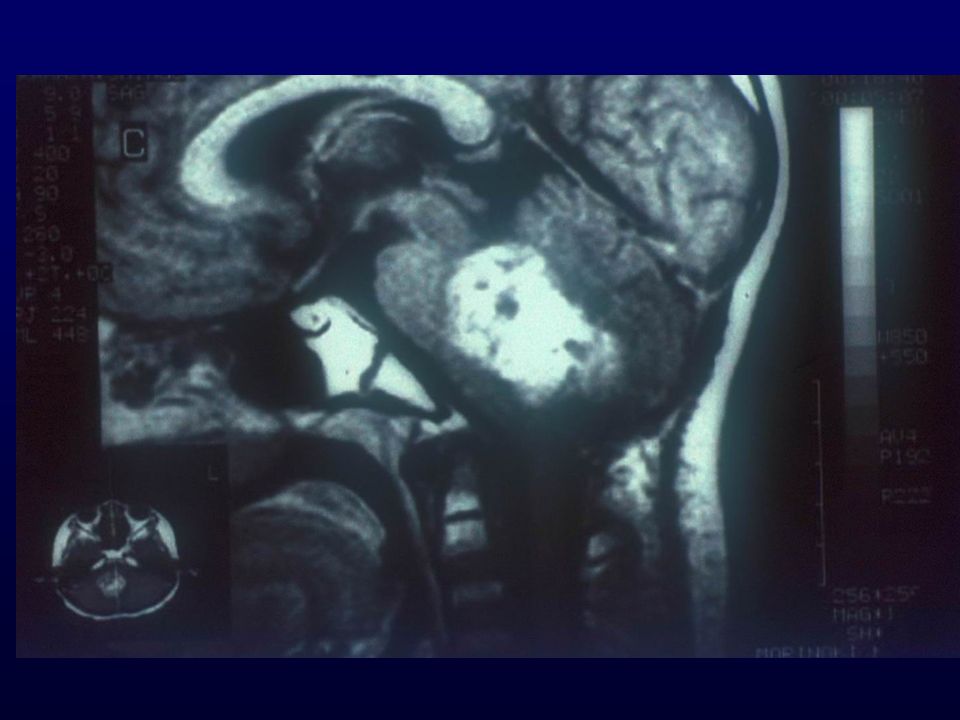
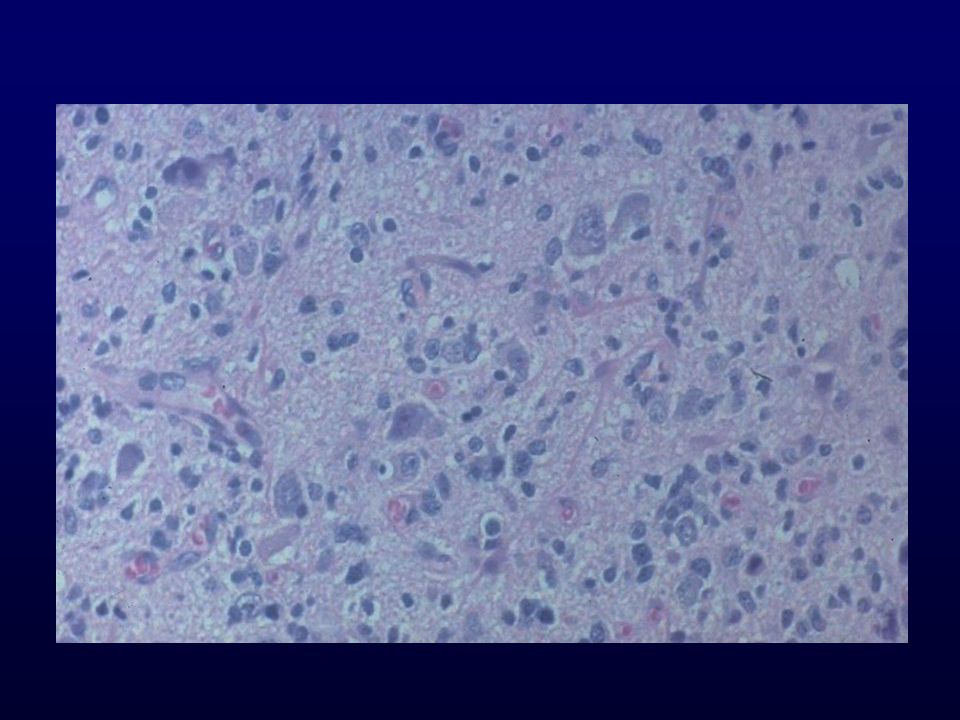
METASTASI CEREBRALE DA ADENOCARCINOMA

Presentazioni simili


















>")








