Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
ESPRESSIONE E PURIFICAZIONE DI UNA PROTEINA DI FUSIONE

2
PRODUZIONE DI PROTEINE PER MEZZO DELL’ INGEGNERIA GENETICA
Applicazioni delle proteine ricombinanti -PROTEINE DI INTERESSE TERAPEUTICO (anticorpi, insulina, ormone crescita). -PROTEINE DI INTERESSE COMMERCIALE (enzimi). -PROTEINE DA UTILIZZARE COME ANTIGENI PER LA PRODUZIONE DI ANTICORPI POLICLONALI E MONOCLONALI. -REAGENTI PER LA RICERCA DI BASE E APPLICATA. Una delle più utili applicazioni della tecnologia del DNA ricombinante è la capacità di sintetizzare artificialmente grandi quantità di proteine naturali o modificate in una cellula ospite (batteri o lieviti), i vantaggi di tali tecniche sono apprezzati da quando nel 1982 furono clonate ed esprese le prime molecole di insulina.
. -PROTEINE DI INTERESSE COMMERCIALE (enzimi). -PROTEINE DA UTILIZZARE COME ANTIGENI PER LA PRODUZIONE DI ANTICORPI POLICLONALI E MONOCLONALI. -REAGENTI PER LA RICERCA DI BASE E APPLICATA. Una delle più utili applicazioni della tecnologia del DNA ricombinante è la capacità di sintetizzare artificialmente grandi quantità di proteine naturali o modificate in una cellula ospite (batteri o lieviti), i vantaggi di tali tecniche sono apprezzati da quando nel 1982 furono clonate ed esprese le prime molecole di insulina.")
3
Sistemi di espressione
Procariotici (E.Coli) Eucariotici: Saccharomyces Cerevisiae (lievito) Cellule di insetto (Baculovirus) Cellule di mammifero in coltura (CHO etc.) Animali transgenici Piante transgeniche
 Eucariotici: Saccharomyces Cerevisiae (lievito) Cellule di insetto (Baculovirus) Cellule di mammifero in coltura (CHO etc.) Animali transgenici. Piante transgeniche.")
4
Anticorpo monoclonale specifico
PROTEINE DI FUSIONE Per evitare degradazione di proteine eterologhe e per permetterne una più semplice purificazione, queste vengono prodotte come proteine di fusione con una proteina stabile dell’organismo ospite. TAG Un TAG è una sequenza proteica con proprietà che ci permettono di purificare la proteina d’interesse TAG DIMENSIONE LIGANDO GST 25 kDa glutatione MBP 40 kDa amilosio FLAG (DYKDDDDK) 8 aa Anticorpo monoclonale specifico His-tag 6-10 aa Ni2+ Le proteine prodotte in un organismo ospite possono essere facilmente soggette a degradazione….
 8 aa. Anticorpo monoclonale specifico. His-tag aa. Ni2+ Le proteine prodotte in un organismo ospite possono essere facilmente soggette a degradazione….")
5
β-Lattamasi GST Ci sono vettori di clonaggio specializzati per l’espressione di geni estranei. i due cdna devono essere fusi mantenendo la corretta cornice di lettura

6
Dominio catalitico PTP1B
PROTEINE DI FUSIONE Dominio catalitico PTP1B PTP GST Le proteine di fusione si costruiscono a livello del DNA saldando le regioni codificanti i due geni

7
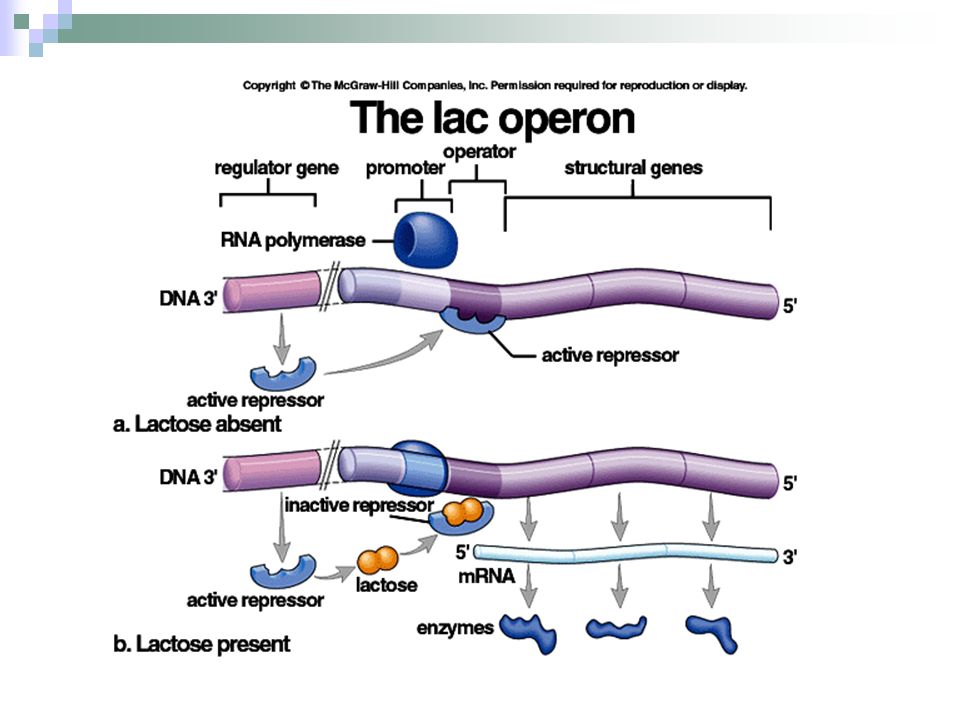
– Alta affinità per l’RNA polimerasi
NELLA PRODUZIONE DI PROTEINE ETEROLOGHE IN BATTERI VENGONO UTILIZZATI SPESSO PROMOTORI FORTI E REGOLABILI Forti – Alta affinità per l’RNA polimerasi – Legame forte – trascritto ad alta frequenza – Legame debole- RNA Pol si stacca, no trasc. Regolabile – L’espressione può essere controllata dal ricercatore, tramite induttori e repressori Il requisito minimo di un sistema di espressione genica efficace è la presenza a monte del gene clonato , di una sequenza promotrice forte regolabile. Un promotore forte ha alta affinità per la rna polimerasi assicurando un’elevata trascrizione della regione a valle. La capacità di regolare il promotore ci dà la possibilità di controllare il grado di trascrizione con precisione. L’operone lac è uno dei più utilizzati UNA PRODUZIONE CONTINUA DELLA PROTEINA PROVOCA: -inibizione funzioni cellula -perdita energia -perdita plasmide

9
Lattosio IPTG

10
ESPRESSIONE E PURIFICAZIONE DI UNA PROTEINA DI FUSIONE
TRASFORMAZIONE DEL VETTORE D’ESPRESSIONE (CODIFICANTE PER LA PROTEINA DI FUSIONE) IN OSPITE. AMPLIFICAZIONE DEL CEPPO BATTERICO TRASFORMATO. INDUZIONE DELL’ESPRESSIONE DELLA PROTEINA DI FUSIONE. PURIFICAZIONE DELLA PROTEINA DI FUSIONE.
 IN OSPITE. AMPLIFICAZIONE DEL CEPPO BATTERICO TRASFORMATO. INDUZIONE DELL’ESPRESSIONE DELLA PROTEINA DI FUSIONE. PURIFICAZIONE DELLA PROTEINA DI FUSIONE.")
11
Assunzione di DNA da parte di un organismo
1. TRASFORMAZIONE Assunzione di DNA da parte di un organismo Amp Gene di interesse E. Coli

12
1. TRASFORMAZIONE

13
2. AMPLIFICAZIONE 12-16 ore 37°C 2-5x109 cellule (N = N02n) LB: Lysogeny Broth (Luria Broth) Tryptone (fonte aminoacidica) Estratto di lievito (vitamine e elementi in tracce) NaCl
 Estratto di lievito (vitamine e elementi in tracce) NaCl.")
14
Unità formanti colonie
latenza esponenziale plateau morte FASE DI LATENZA: intervallo di tempo in cui i microrganismi si adattano al substrato (non si moltiplicano) FASE ESPONENZIALE: periodo di tempo in cui i microrganismi si moltiplicano in modo progressivo e costante PLATEAU: si stabilisce un equilibrio tra il numero delle cellule che si moltiplicano e il numero di cellule che muoiono FASE DELLA MORTE: cessa la moltiplicazione dei microrganismi che invecchiano e muoiono
 FASE ESPONENZIALE: periodo di tempo in cui i microrganismi si moltiplicano in modo progressivo e costante. PLATEAU: si stabilisce un equilibrio tra il numero delle cellule che si moltiplicano e il numero di cellule che muoiono. FASE DELLA MORTE: cessa la moltiplicazione dei microrganismi che invecchiano e muoiono.")
15
3. INDUZIONE assorbanza ore DILUIZIONE IPTG 2 ore 1:100 37°C

16
LISI BATTERICA ED ESTRAZIONE DELLA COMPONENTE PROTEICA
4. PURIFICAZIONE LISI BATTERICA ED ESTRAZIONE DELLA COMPONENTE PROTEICA supernatante 10’ 1. Recupero batteri Pellet (batteri) 4000 rpm congelamento/scongelamento Lisozima (1 mg/ml) (agisce sulla membrana esterna) 2. Lisi Sonicazione (ultrasuoni) detergenti Pellet (membrane, DNA) 30’ Supernatante (proteine) 3. Recupero componente proteica 13000 rpm
 4000 rpm. congelamento/scongelamento. Lisozima (1 mg/ml) (agisce sulla membrana esterna) 2. Lisi. Sonicazione (ultrasuoni) detergenti. Pellet (membrane, DNA) 30’ Supernatante. (proteine) 3. Recupero componente proteica rpm.")
17
AZIONE DEL LISOZIMA PARETE Gram-
Il lisozima è un enzima di 14,4 kDa presente in tessuti animali dotato di attività battericida. Lisa la parete batterica di alcuni batteri catalizzando l'idrolisi del legame beta 1,4 tra l’acido N-acetilmuramico (NAM) e la N-acetilglucosamina (NAG) che sono la componente principale delpeptidoglicano. PARETE Gram- LISOZIMA
 e la N-acetilglucosamina (NAG) che sono la componente principale delpeptidoglicano. PARETE Gram- LISOZIMA.")
18
Immobilizzazione della proteina di fusione su supporto solido
ESTRATTO PROTEICO NaCl KCl Na2HPO4 KH2PO4 GLUTATIONE IMMOBILIZZATO SU BIGLIE DI SEFAROSIO RIMUOVO IL SUPERNATANTE CONTENENTE LE PROTEINE NON LEGATE 3000 RPM + LAVAGGI con PBS

19
5. VERIFICA TRAMITE CORSA ELETTROFORETICA
SDS-PAGE (Sodium Dodecyl (lauryl) Sulfate PolyAcrylamide Gel Electrophoresis) - POZZETTI PER CARICAMENTO TRIS-HCl 0.5 M pH 6.8 ACRILAMIDE 5% SDS STACKING GEL TRIS-HCl 1.5 M pH 8.8 ACRILAMIDE > 5% SDS La nostra proteina di fusione deve migrare all’altezza corrispondente al suo peso molecolare SEPARATING GEL +
 Sulfate PolyAcrylamide Gel Electrophoresis) - POZZETTI PER CARICAMENTO. TRIS-HCl 0.5 M pH 6.8. ACRILAMIDE 5% SDS. STACKING GEL. TRIS-HCl 1.5 M pH 8.8. ACRILAMIDE > 5% SDS. La nostra proteina di fusione deve migrare all’altezza corrispondente al suo peso molecolare. SEPARATING GEL. +")
20
stacking gel: è la parte superiore del gel e la sua funzione è quella di concentrare il campione proteico caricato negli appositi pozzetti, in modo che tutti i campioni comincino la loro migrazione dallo stesso punto di partenza. running gel: è la parte inferiore e la sua funzione è quella di separare le proteine dei vari campioni sulla base del loro peso molecolare. È composto dagli stessi ingredienti dello stacking gel, ma in quantità diverse. In particolare è la concentrazione di acrilammide a variare, a seconda della porosità desiderata: concentrazioni maggiori portano a pori di dimensioni minori, dunque capaci di separare le proteine con una risoluzione maggiore.

21
Loading buffer COME PREPARARE IL CAMPIONE PER LA CORSA loading buffer
+ + 95° C 13000 rpm L’ELEVATA TEMPERATURA ROMPE IL LEGAME TRA GST E BIGLIE DI SEFAROSIO L’SDS DENATURA LA PROTEINA E ROMPE LE INTERAZIONI IL BETA-MERCAPTOETANOLO RIDUCE GLI EVENTUALI PONTI DISOLFURO CENTRIFUGANDO POSSIAMO ISOLARE IL SUPERNATANTE CONTENENTE LA PROTEINA Loading buffer 50 mM Tris-HCl pH 6.8 2% SDS 10% Glycerol 1% b-Mercaptoethanol mM EDTA % Bromophenol Blue

22
RUOLO DELL’SDS NELLA CORSA
LE CARICHE E LA FORMA DI UNA PROTEINA NATIVA INFLUISCONO SULLA CORSA PROTEINA NATIVA L’SDS LEGA LE PROTEINE IN UN RAPPORTO DI UN ANIONE OGNI 2 aa PERDITA DELLE STRUTTURE SECONDARIA E TERZIARIA ACQUISIZIONE DI UNA CARICA NETTA NEGATIVA RAPPORTO CARICA/MASSA SIMILE PER TUTTE LE PROTEINE NEL CAMPIONE Il trattamento con SDS elimina le differenze di forma, quindi la lunghezza della catena polipeptidica, che riflette la massa, e’ l’unico determinante della velocita’ di migrazione. La separazione avviene quindi per differenza fra pesi molecolari visto che il rapporto massa carica per ogni proteina denaturata con SDS rimane costante.

23
25 mM Tris HCl pH 8.3 192 mM Glicina 0.1% SDS
IL GEL VA IMMERSO IN UNA SOLUZIONE DI CORSA CONTENENTE GLICINA 25 mM Tris HCl pH 8.3 192 mM Glicina 0.1% SDS

24
Nel buffer a pH 8.3 la glicina è carica negativamente e comincia
a migrare con una certa velocità Arrivata al loading buffer e allo stacking (entrambi a pH 6.8) la glicina è quasi neutra ed è la specie più lenta, mentre gli ioni Cl- sono la specie più veloce Gly Gly Gly Gly pH 8.3 Cl- Cl- pH 6.8 Cl- Cl- Cl- Cl- pH 8.8 Cl- Cl- Cl- Questo fa sì che le proteine vengano racchiuse in una regione ad elevata mobilità elettroforetica
 la glicina è quasi neutra. ed è la specie più lenta, mentre gli ioni Cl- sono la specie più veloce. Gly. Gly. Gly. Gly. pH 8.3. Cl- Cl- pH 6.8. Cl- Cl- Cl- Cl- pH 8.8. Cl- Cl- Cl- Questo fa sì che le proteine. vengano racchiuse in una regione. ad elevata mobilità elettroforetica.")
25
del campione in base al peso molecolare.
EFFETTO STACKING Gly PROTEINE COLORANTE Cl- Cl- Cl- Cl- All’inizio del separating gel il pH 8.8 riporta la glicina ad un rapporto carica/massa maggiore rispetto a quello delle proteine. La glicina supera i polipeptidi, che non trovandosi più in una regione ad elevata mobilità elettroforetica rallentano notevolmente e si ritrovano compattati in una linea molto sottile. Ora le proteine si trovano tutte allo stesso livello. Nel separating la concentrazione di acrilamide è più alta, e comincia la separazione del campione in base al peso molecolare.

26
Alla fine della corsa il gel può essere colorato con blu Coomassie
Alla fine della corsa il gel può essere colorato con blu Coomassie. Il colorante lega arginina, istidina e aminoacidi aromatici, rendendo visibili le proteine su gel in base alla loro quantità.

27
APPLICAZIONI SI PUO’ SFRUTTARE IL SUPPORTO SOLIDO PER ESPERIMENTI DI PULL DOWN SI PUO’ ELUIRE LA PROTEINA LEGATA DALLA RESINA DI SEFAROSIO E UTILIZZARLA PER SAGGI ENZIMATICI 3. SI PUO’ SFRUTTARE IL PRINCIPIO DEL TAG PER ESPERIMENTI DI DOPPIO IBRIDO

Presentazioni simili















polimeri è quello della determinazione del peso molecolare. I polimeri.>")

 sottoposte a un campo elelttrico.>")

