Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Chimica Elettroanalitica Avanzata Modulo A
Corso di Laurea Magistrale in Scienze Chimiche Chimica Elettroanalitica Avanzata Modulo A Conduttimetria Prof. Patrizia R. Mussini Dipartimento di Chimica Fisica ed Elettrochimica Via Golgi 19, Milano

2
in soluzione e in membrana
Richiami sui fenomeni di trasporto in soluzione e in membrana

3
In questa sezione ci soffermiamo sulla migrazione elettrica
A1. Meccanismi di trasporto in soluzione Il trasporto di una specie in soluzione avviene attraverso tre meccanismi: diffusione (driving force: un gradiente di potenziale chimico, cioè di concentrazione) In questa sezione ci soffermiamo sulla migrazione elettrica e sulla grandezza ad essa correlata, la conduttività (o “conduttanza specifica”) delle soluzioni elettrolitiche migrazione elettrica (driving force: un gradiente di potenziale elettrico ) convezione (driving forces: temperatura, agitazione, ecc.) I parametri chiave di migrazione e diffusione, ovvero rispettivamente le mobilità ioniche ui e i coefficienti di diffusione degli stessi ioni Di, sono fra di loro collegati (dall’importante equazione di Nernst-Einstein) Di = (RT/zF) ui
 In questa sezione ci soffermiamo sulla migrazione elettrica. e sulla grandezza ad essa correlata, la conduttività (o conduttanza specifica ) delle soluzioni elettrolitiche. migrazione elettrica (driving force: un gradiente di potenziale elettrico ) convezione (driving forces: temperatura, agitazione, ecc.) I parametri chiave di migrazione e diffusione, ovvero rispettivamente le mobilità ioniche ui e i coefficienti di diffusione degli stessi ioni Di, sono fra di loro collegati (dall’importante equazione di Nernst-Einstein) Di = (RT/zF) ui.")
4
Soluzioni elettrolitiche
A2. Un paragone di conduttività tra conduttori elettronici e ionici Metalli Semimetallo Sale fuso Elettrolita solido Soluzioni elettrolitiche Semiconduttore Isolante

5
A3. Conduttività dal punto di vista macroscopico/sperimentale
Dal punto di vista sperimentale un conduttimetro misura la resistenza R () di una soluzione elettrolitica, il cui inverso è la conduttanza G (S[iemens]= -1); essa dipende non solo dalle caratteristiche della soluzione, ma anche da quelle della cella di conduttività usata per effettuare la misura, rappresentate dalla costante di cella (cm-1) (dove, nel caso teorico di una cella per conduttività ideale con linee di corrente rettilinee e parallele, L= distanza tra le piastre in cm e S =superficie delle piastre in cm2). Mediante l’espressione conoscendo il valore della costante di cella, si passa al parametro conduttività o conduttanza specifica (-1 cm-1), che rappresenta le proprietà di trasporto globali di una soluzione elettrolitica La conduttività delle soluzioni viene misurata in condizioni di corrente alternata in una cella simmetrica a due elettrodi
 di una soluzione elettrolitica, il cui inverso è la conduttanza G (S[iemens]= -1); essa dipende non solo dalle caratteristiche della soluzione, ma anche da quelle della cella di conduttività usata per effettuare la misura, rappresentate dalla costante di cella (cm-1) (dove, nel caso teorico di una cella per conduttività ideale con linee di corrente rettilinee e parallele, L= distanza tra le piastre in cm e S =superficie delle piastre in cm2). Mediante l’espressione. conoscendo il valore della costante di cella, si passa al parametro conduttività o conduttanza specifica (-1 cm-1), che rappresenta le proprietà di trasporto globali di una soluzione elettrolitica. La conduttività delle soluzioni viene misurata in condizioni di corrente alternata in una cella simmetrica a due elettrodi.")
6
A4. Conduttività dal punto di vista microscopico
Carica ione Concentrazione ione Mobilità ione mobilità ui = velocità dello ione i sotto gradiente di potenziale unitario, espressa in (cm/s)/(V/cm) = cm2/(V s) concentrazione ci solo qui, in mol/cm3 Verifica dimensionale: (mol e/mol)•(mol/cm3) •(cm/s)/(V/cm)•(C/mol e) = C/(s V cm) = A /(V cm) = W-1 cm-1 (Dunque coincide con secondo l’approccio macroscopico) La conduttività è una grandezza intrinsecamente non selettiva ma “integrale” perché dipende dai prodotti (carica × concentrazione × mobilità) di tutti gli ioni liberi nella soluzione studiata
/(V/cm) = cm2/(V s) concentrazione ci. solo qui, in mol/cm3. Verifica dimensionale: (mol e/mol)•(mol/cm3) •(cm/s)/(V/cm)•(C/mol e) = C/(s V cm) = A /(V cm) = W-1 cm-1 (Dunque coincide con secondo l’approccio macroscopico) La conduttività è una grandezza intrinsecamente non selettiva ma integrale perché dipende dai prodotti (carica × concentrazione × mobilità) di tutti gli ioni liberi nella soluzione studiata.")
7
H+ ui = |zi|e / (6 °ri.ST) Altri ioni: “Moto viscoso” [Stokes]
A5. Meccanismi di moto degli ioni a confronto Altri ioni: “Moto viscoso” [Stokes] Ione H+ : “Moto a salto” [Grotthus] Movimento lento che decresce all’aumentare della viscosità del solvente. Movimento quasi istantaneo H+ Se i siti d’acqua a disposizione calano, cala la probabilità che i protoni si muovano col meccanismo a salto e la loro mobilità cala In mezzo aprotico anche i protoni hanno mobilità simili agli altri ioni Legge di Stokes: ui = |zi|e / (6 °ri.ST) Come nel biliardo: La mobilità: cala all’aumentare della viscosità del solvente cala all’aumentare del raggio ionico SOLVATATO (Cs+ < Rb+ < K+ < Na+ < Li+) Specifico per gli ioni H+, richiede disponibilità di un sufficiente numero di siti H2O
![H+ ui = |zi|e / (6 °ri.ST) Altri ioni: Moto viscoso [Stokes]](http://slideplayer.it/slide/571549/1/images/7/H%2B+ui+%3D+%7Czi%7Ce+%2F+%286%EF%81%B0+%EF%81%A8%C2%B0ri.ST%29+Altri+ioni%3A+Moto+viscoso+%5BStokes%5D.jpg "A5. Meccanismi di moto degli ioni a confronto. Altri ioni: Moto viscoso [Stokes] Ione H+ : Moto a salto [Grotthus] Movimento lento che decresce all’aumentare della viscosità del solvente. Movimento quasi istantaneo. H+ Se i siti d’acqua a disposizione calano, cala la probabilità che i protoni si muovano col meccanismo a salto e la loro mobilità cala. In mezzo aprotico anche i protoni hanno mobilità simili agli altri ioni. Legge di Stokes: ui = |zi|e / (6 °ri.ST) Come nel biliardo: La mobilità: cala all’aumentare della viscosità del solvente. cala all’aumentare del raggio ionico SOLVATATO (Cs+ < Rb+ < K+ < Na+ < Li+) Specifico per gli ioni H+, richiede disponibilità di un sufficiente numero di siti H2O.")
8
HCl K2SO4 KCl CH3COOH A6. Influenza della concentrazione
a concentrazione c Per HCl F = costante di Faraday =96485 C mol e-1 = grado di dissociazione dipende dalla concentrazione sia direttamente sia indirettamente tramite ed u. In particolare si ha: elettroliti forti: la curva vs c presenta un massimo (benché non sempre raggiunto, poichè all’inizio predomina la crescita di c(tratto ascendente della curva), poi il calo di ed u con la concentrazione ( tratto discendente della curva); elettroliti deboli: le mobilità sono circa costanti, la crescita della concentrazione e del grado di dissociazione si compensano per cui la curva vs c ha un andamento pianeggiante. K2SO4 KCl CH3COOH
, poi il calo di ed u con la concentrazione ( tratto discendente della curva); elettroliti deboli: le mobilità sono circa costanti, la crescita della concentrazione e del grado di dissociazione si compensano per cui la curva vs c ha un andamento pianeggiante. K2SO4. KCl. CH3COOH.")
9
Accumulatore al piombo:
L’esempio dell’acido solforico 21 °C 33.5% M V 71 °C 82 °C PbO2 + Pb + 2 H2SO4 = 2 PbSO4 + 2 H2O scarica carica Accumulatore al piombo:

10
conduttanza / W-1 = f (soluzione, cella)
A7. Parametri che descrivono le proprietà di trasporto di una soluzione elettrolitica in modo assoluto conduttanza / W-1 = f (soluzione, cella) Normalizzazione per la costante di cella conduttanza specifica o conduttività / (W-1 cm-1) = f (soluzione) Normalizzazione per la concentrazione conduttanza molare / (W-1 mol-1 cm2) Approssimazioni per soluzioni molto diluite: 1) Elettroliti forti: ) Elettroliti deboli CA = C+ + A- K = c2/(1-) = u circa costante, =/° Equazione di Onsager Equazione di Kraus e Bray a0=ion-size parameter in cm, B= •108 dm3/2 mol-1/2cm-1 B1= (mol/dm3)-1/2 B2= cm2 -1 mol-3/2dm-3/2 (a T=25°C)
 Normalizzazione per la costante di cella. conduttanza specifica o conduttività / (W-1 cm-1) = f (soluzione) Normalizzazione per la concentrazione. conduttanza molare. / (W-1 mol-1 cm2) Approssimazioni per soluzioni molto diluite: 1) Elettroliti forti: 2) Elettroliti deboli CA = C+ + A- K = c2/(1-) = 1 u circa costante, =/° Equazione di Onsager. Equazione di Kraus e Bray. a0=ion-size parameter in cm, B= •108 dm3/2 mol-1/2cm-1. B1= (mol/dm3)-1/2. B2= cm2 -1 mol-3/2dm-3/2. (a T=25°C)")
11
A8. Conduttività molari a diluizione infinita
e legge di Kohlrausch della migrazione indipendente degli ioni dipende ancora dalla concentrazione ma solo implicitamente, tramite ed u. A diluizione infinita = 1 conduttività ioniche molari limite, sono caratteristiche di ciascuno ione ed indipendenti dall’elettrolita di partenza (legge di Kohlrausch della migrazione indipendente degli ioni) per questo, la conduttività molare ° a diluizione infinita di un elettrolita si può ricavare per combinazione di dati di ° relativi ad altri elettroliti o di singoli ioni. Si noti anche che °+= z+F u° °= zF u° e poiché Di = (RT/zF) ui Di°+ = RT/(zF)2 °+ Di° = RT/(zF)2 °
 per questo, la conduttività molare ° a diluizione infinita di un elettrolita. si può ricavare per combinazione di dati di ° relativi ad altri elettroliti o di singoli ioni. Si noti anche che °+= z+F u°+ °= zF u° e poiché Di = (RT/zF) ui. Di°+ = RT/(zF)2 °+ Di° = RT/(zF)2 °")
12
Conduttività molari cationiche e anioniche,°+ e°– ( cm2 mol-1) a diluizione infinita in acqua
Giustificate le conduttività di H+ e OH- nei confronti degli altri ioni Giustificate le sequenze di conduttività all’interno dei cationi alcalini, dei cationi alcalino terrosi e degli alogenuri. Se l’acqua ha viscosità 1 cP mentre metanolo e propilencarbonato hanno viscosità cP e 2.53 cP rispettivamente, valutate la mobilità dello ione Cs+ in tali solventi. Potreste fare lo stesso calcolo con H+? E con Li+? Perché? Calcolate °CaCl2 Sapendo che °KCl = , °HCl = , °K2SO4 = calcolate °H2SO4 Calcolate i coefficienti di diffusione degli ioni Na+ e Cl– a diluizione infinita. R. A. Robinson, R. H. Stokes, "Electrolyte Solutions", 1965, Butterworths, London, p. 463

13
A9. Verifica dell’equazione di Onsager per gli elettroliti forti
°HCl °K2SO4 °KCl Conduttività molari a diluizione infinita

14
A10. Dimostrazione dell’equazione di Kraus e Bray
Hp per elettrolita debole: mobilità circa costanti (il numero di particelle è sempre molto basso) costante di dissociazione di un elettrolita debole, ad esempio uniunivalente, in funzione del grado di dissociazione, trascurando i coefficienti di attività yi
 costante di dissociazione di un elettrolita debole, ad esempio uniunivalente, in funzione del grado di dissociazione, trascurando i coefficienti di attività yi.")
15
A11. Verifica dell’equazione di Kraus e Bray
CH3COOH 1/°

16
A12. Condizioni per la misura della conduttività di una soluzione
La misura di conduttività di una soluzione elettrolitica si esegue in corrente alternata (normalmente alla frequenza di 1000 Hz) in modo da evitare polarizzazione o addirittura processi elettrolitici agli elettrodi. Però una soluzione non è un conduttore puramente ohmico. I circuiti non puramente ohmici percorsi da corrente alternata risentono di effetti induttivi e/o capacitivi che sono funzione della frequenza della corrente impiegata. Cioè, mandando una tensione alternata sulla cella di conduttività e misurando la corrente, la risposta del sistema non è di una resistenza ohmica, ma di una impedenza Z, cioè la somma vettoriale della resistenza ohmica R e di termini resistivi corrispondenti agli elementi capacitivi e induttivi presenti nel circuito, rispettivamente “reattanze capacitive XC ” e “reattanze induttive XL”.
 in modo da evitare polarizzazione o addirittura processi elettrolitici agli elettrodi. Però una soluzione non è un conduttore puramente ohmico. I circuiti non puramente ohmici percorsi da corrente alternata risentono di effetti induttivi e/o capacitivi che sono funzione della frequenza della corrente impiegata. Cioè, mandando una tensione alternata sulla cella di conduttività e misurando la corrente, la risposta del sistema non è di una resistenza ohmica, ma di una impedenza Z, cioè la somma vettoriale della resistenza ohmica R e di termini resistivi corrispondenti agli elementi capacitivi e induttivi presenti nel circuito, rispettivamente reattanze capacitive XC e reattanze induttive XL .")
17
A13. Lo strumento storico: il ponte di Kohlrausch
Poiché si può considerare la cella di conduttività equivalente ad una R e una C in parallelo, lo strumento storico per le misure di resistenza di una soluzione elettrolitica, il ponte di Kohlrausch, permetteva di effettuare la misura bilanciando il parallelo RxCx della cella variando un parallelo campione RcCc fino ad azzerare il modulo e lo sfasamento della corrente alternata nel ramo centrale del ponte.

18
A14. Conduttimetri di routine
Tuttavia i conduttimetri moderni di routine, basati sugli amplificatori operazionali, determinano direttamente la resistenza ohmica della soluzione R (e quindi il suo inverso, la conduttanza G) nel modo concettualmente più semplice, cioè mandando una tensione costante e nota con precisione, e rilevando la corrente, cosa possibile perché contemporaneamente si riesce a minimizzare l’effetto delle componenti capacitive del sistema. Inoltre i conduttimetri moderni di routine: Permettono l’impostazione della costante di cella, con la quale lo strumento calcola e fornisce direttamente la conduttività Permettono la correzione della misura riportandola dalla temperatura attuale ad una temperatura di riferimento Hanno sul retro una uscita che fornisce una differenza di potenziale adatta per riplatinare la cella di misura.
 nel modo concettualmente più semplice, cioè mandando una tensione costante e nota con precisione, e rilevando la corrente, cosa possibile perché contemporaneamente si riesce a minimizzare l’effetto delle componenti capacitive del sistema. Inoltre i conduttimetri moderni di routine: Permettono l’impostazione della costante di cella, con la quale lo strumento calcola e fornisce direttamente la conduttività Permettono la correzione della misura riportandola dalla temperatura attuale ad una temperatura di riferimento. Hanno sul retro una uscita che fornisce una differenza di potenziale adatta per riplatinare la cella di misura.")
19
A15. Celle conduttimetriche
Si chiama cella conduttimetrica l’insieme comprendente gli elettrodi attraverso i quali viene trasmessa al liquido in esame la corrente di misura, le parti isolanti che delimitano la porzione di soluzione percorsa dalla corrente di misura e infine le ulteriori parti, isolanti e non, che servono per l’unione meccanica delle parti principali, per la tenuta ermetica, per il collegamento al circuito esterno, ecc. Spesso si commette l’errore di chiamare “elettrodo” quello che in realtà è una coppia di elettrodi o addirittura una vera e propria cella! Tipica cella da laboratorio con 2 elettrodi di platino, platinato, con parti isolanti e riparo di vetro (che può essere sostituito dalla plastica solo per applicazioni di routine in solvente compatibile). Essi sono preferiti nelle misure di laboratorio, perchè resistono a quasi tutti gli agenti chimici, e anche per la elevata capacità che si ha per l’interfase elettrodo/soluzione (per cui risulta molto bassa, a parità di frequenza operativa, la reattanza capacitiva di tale interfase). Si deve controllare periodicamente lo stato del sottile e delicato strato di nero di platino, e, in caso di danneggiamento, ridepositarlo elettroliticamente con apposito bagno di platinatura; alcuni strumenti prevedono un piccolo alimentatore proprio a questo scopo Per il collegamento con l’apparecchio la cella termina con 2 boccole corrispondenti ai due elettrodi. Esse sono ovviamente del tutto equivalenti, visto che si opera in tensione alternata.
. Essi sono preferiti nelle misure di laboratorio, perchè resistono a quasi tutti gli agenti chimici, e anche per la elevata capacità che si ha per l’interfase elettrodo/soluzione (per cui risulta molto bassa, a parità di frequenza operativa, la reattanza capacitiva di tale interfase). Si deve controllare periodicamente lo stato del sottile e delicato strato di nero di platino, e, in caso di danneggiamento, ridepositarlo elettroliticamente con apposito bagno di platinatura; alcuni strumenti prevedono un piccolo alimentatore proprio a questo scopo. Per il collegamento con l’apparecchio la cella termina con 2 boccole corrispondenti ai due elettrodi. Esse sono ovviamente del tutto equivalenti, visto che si opera in tensione alternata.")
20
Celle a immersione Celle a deflusso
A15. Celle conduttimetriche industriali Industrialmente si usano elettrodi di acciaio inossidabile (o di grafite) con parti isolanti di resina: sono più semplici, robusti e poco costosi, ma con prestazioni inferiori, specie per resistenze molto basse (conduttività molto alte) Celle a immersione Celle a deflusso particolarmente utili, montati su bypass delle tubazioni principali, per controlli industriali in continuo.
 con parti isolanti di resina: sono più semplici, robusti e poco costosi, ma con prestazioni inferiori, specie per resistenze molto basse (conduttività molto alte) Celle a immersione. Celle a deflusso. particolarmente utili, montati su bypass delle tubazioni principali, per controlli industriali in continuo.")
21
A16. Determinazione della costante di cella
Qualsiasi cella conduttimetrica è caratterizzata dalla propria costante di cella L/S (in cm1) o S/L (in cm), e, nel caso di forme geometriche semplici, teoricamente potrebbe essere calcolata dalla lunghezza L e sezione attiva S del conduttore, mentre nel caso di forme irregolari, si dovrebbe sviluppare un integrale. Però, soprattutto a causa della geometria non ideale delle linee di corrente e dell’uso di superfici conduttrici complesse è molto meglio operare una taratura misurando la G di una soluzione campione di cui è nota con precisione la e utilizzando la /G = (L/S).
 o S/L (in cm), e, nel caso di forme geometriche semplici, teoricamente potrebbe essere calcolata dalla lunghezza L e sezione attiva S del conduttore, mentre nel caso di forme irregolari, si dovrebbe sviluppare un integrale. Però, soprattutto a causa della geometria non ideale delle linee di corrente e dell’uso di superfici conduttrici complesse è molto meglio operare una taratura misurando la G di una soluzione campione di cui è nota con precisione la e utilizzando la /G = (L/S).")
22
A17. Standardizzazione primaria (I)
Tradizionalmente la standardizzazione primaria veniva effettuata con mercurio, metallo liquido di cui è nota la conduttività con grandissima precisione. La strategia seguita si può così riassumere (in modo molto semplificato): Con Hg determino la costante di cella di una cella con un’alta costante di cella, data la conduttività elevatissima del mercurio, circa volte più alta di quella di KCl 1 M usato come standard di laboratorio: [considerando la equazione R = 1/G = 1/ (L/S) si può mantenere entro il range strumentale ottimale la misura di conduttanza G di soluzioni di anche molto diversa, utilizzando celle con L/S alta per alte, bassa per basse] 1/G1 = 1/ Hg (L/S)1 Successivamente nella medesima cella si determina la conduttività di una soluzione concentrata di H2SO4. 1/G2 = 1/ H2SO4 (L/S)1 Con questa soluzione intermedia si effettua la taratura di una seconda cella con costante L/S più bassa, 1/G3 = 1/ H2SO4 (L/S)2 con la quale si determina infine la conduttività delle soluzioni di KCl. 1/G4 = 1/ KCl (L/S)2
: Con Hg determino la costante di cella di una cella con un’alta costante di cella, data la conduttività elevatissima del mercurio, circa volte più alta di quella di KCl 1 M usato come standard di laboratorio: [considerando la equazione R = 1/G = 1/ (L/S) si può mantenere entro il range strumentale ottimale la misura di conduttanza G di soluzioni di anche molto diversa, utilizzando celle con L/S alta per alte, bassa per basse] 1/G1 = 1/ Hg (L/S)1. Successivamente nella medesima cella si determina la conduttività di una soluzione concentrata di H2SO4. 1/G2 = 1/ H2SO4 (L/S)1. Con questa soluzione intermedia si effettua la taratura di una seconda cella con costante L/S più bassa, 1/G3 = 1/ H2SO4 (L/S)2. con la quale si determina infine la conduttività delle soluzioni di KCl. 1/G4 = 1/ KCl (L/S)2.")
23
A18. Standardizzazione primaria (II)
Attualmente il National Institute of Standards and Technology USA NIST sta impiegando, per la determinazione diretta di standard primari KCl 0.1 M e KCl 0.01 M, un metodo geometrico differenziale con il modulo di espansione senza il modulo di espansione

24
A19. Taratura del conduttimetro nella routine di laboratorio (I)
Nella routine di laboratorio si richiede però solo la frequente rideterminazione della costante di cella (che può variare con il tempo e l’uso rispetto a quella indicata dal commerciante) mediante la misura della conduttanza a temperatura nota di uno standard operativo adatto alla cella da usarsi e di conduttività nota con precisione. Per le celle di uso corrente (con costante L/S 1) gli standard correntemente usati sono soluzioni di KCl 0.1 m o 0.01 m. Assai a proposito qui, come altrove, nella definizione di standard si fa uso della molalità (moli soluto/kg solvente) invece della molarità (moli soluto/dm3 soluzione). Infatti la prima implica di preparare le soluzioni interamente per pesata e quindi può raggiungere una precisione molto più elevata; a differenza della seconda, non è influenzata dalla temperatura. Le soluzioni di KCl per la taratura dovrebbero essere preparate utilizzando: KCl purissimo per analisi essiccato in stufa ( ° per almeno 2 ore) e lasciato raffreddare in essiccatore acqua bidistillata o deionizzata bollita di fresco per eliminare la CO2 e raffreddata in recipiente protetto con tubo di calce sodata, di conduttività trascurabile a quella della soluzione standard che si vuole preparare, e comunque 1-2 mS/cm a 25 °C; durante la preparazione non va esposta all’aria per evitare assorbimento di CO2 ed altre impurezze. Le soluzioni standard preparate vanno anch’esse protette con tubo a calce sodata e hanno validità limitata (alcuni mesi).
 mediante la misura della conduttanza a temperatura nota di uno standard operativo adatto alla cella da usarsi e di conduttività nota con precisione. Per le celle di uso corrente (con costante L/S 1) gli standard correntemente usati sono soluzioni di KCl 0.1 m o 0.01 m. Assai a proposito qui, come altrove, nella definizione di standard si fa uso. della molalità (moli soluto/kg solvente) invece della molarità (moli soluto/dm3 soluzione). Infatti la prima implica di preparare le soluzioni interamente per pesata e quindi. può raggiungere una precisione molto più elevata; a differenza della seconda, non è influenzata dalla temperatura. Le soluzioni di KCl per la taratura dovrebbero essere preparate utilizzando: KCl purissimo per analisi essiccato in stufa ( ° per almeno 2 ore) e lasciato raffreddare in essiccatore. acqua bidistillata o deionizzata bollita di fresco per eliminare la CO2 e raffreddata in recipiente protetto con tubo di calce sodata, di conduttività trascurabile a quella della soluzione standard che si vuole preparare, e comunque 1-2 mS/cm a 25 °C; durante la preparazione non va esposta all’aria per evitare assorbimento di CO2 ed altre impurezze. Le soluzioni standard preparate vanno anch’esse protette con tubo a calce sodata e hanno validità limitata (alcuni mesi).")
25
A19. Taratura del conduttimetro nella routine di laboratorio
Per le soluzioni standard KCl 0.1 m e 0.01 m il National Institute of Standards and Technology USA raccomanda i seguenti valori di conduttività, tabulati in funzione della temperatura: The values in Table 1 were corrected for the electrolytic conductivity of the solvent, i.e., water in equilibrium with atmospheric CO2, at the temperature of measurement. The measured conductivities of the solvent are given in the rightmost column of Table 1. This solvent conductivity is subtracted from the value measured for the test solution (not shown) to yield the reported conductivity, κ, at the given molality and Celsius temperature.
 to yield the reported conductivity, κ, at the given molality and Celsius temperature.")
26
A20. Correzione della temperatura
Il controllo della temperatura nelle misure di conduttività è estremamente importante, perchè la conduttività varia sensibilmente con la temperatura. Una equazione empirica ad hoc è la seguente: = rif (1+ (T Trif) + (T Trif)2 +...) Quesito. Sapendo che 0.02 K-1, con quale precisione occorre termostatare la soluzione di misura in modo da contenere l’errore relativo entro lo 0.1%? Considerando solo i primi due termini della polinomiale in parentesi: = rif (1+(T Trif)) |rif|/rif = errore relativo = (T Trif) 0.001 (T Trif) = errore assoluto sulla temperatura = 0.001/0.02 0.05 Quindi per contenere l’errore relativo sulla conduttività entro lo 0.1% occorre termostatare con la precisione di mezzo decimo di grado. Se non è possibile termostatare le soluzioni alla temperatura di riferimento a cui si possiedono dati tabulati (ad esempio 25°C), è possibile, conoscendo il valore della temperatura attuale, riportare le misure alla temperatura di riferimento con un calcolo, mediante una polinomiale quale quella dell’esempio o quelle delle Tabelle citate. Molti conduttimetri sono in grado di effettuare direttamente questa correzione, purchè si forniscano loro le seguenti informazioni: 1) temperatura di riferimento (ora più spesso 25°C, una volta 18°C); 2) temperatura attuale (che può essere rilevata automaticamente da una termosonda, o letta dallo sperimentatore ed inserita manualmente); 3) valore del coefficiente di temperatura (noto a priori o determinabile al momento), con l’assunzione che l’intervallo tra T attuale e T riferimento sia sufficientemente piccolo da poter stimare lineare l’incremento della conduttività con la temperatura.
 + (T Trif)2 +...) Quesito. Sapendo che 0.02 K-1, con quale precisione occorre termostatare la soluzione di misura. in modo da contenere l’errore relativo entro lo 0.1% Considerando solo i primi due termini della polinomiale in parentesi: = rif (1+(T Trif)) |rif|/rif = errore relativo = (T Trif) (T Trif) = errore assoluto sulla temperatura = 0.001/0.02 Quindi per contenere l’errore relativo sulla conduttività entro lo 0.1% occorre termostatare con la precisione di mezzo decimo di grado. Se non è possibile termostatare le soluzioni alla temperatura di riferimento a cui si possiedono dati tabulati (ad esempio 25°C), è possibile, conoscendo il valore della temperatura attuale, riportare le misure alla temperatura di riferimento con un calcolo, mediante una polinomiale quale quella dell’esempio o quelle delle Tabelle citate. Molti conduttimetri sono in grado di effettuare direttamente questa correzione, purchè si forniscano loro le seguenti informazioni: 1) temperatura di riferimento (ora più spesso 25°C, una volta 18°C); 2) temperatura attuale (che può essere rilevata automaticamente da una termosonda, o letta dallo sperimentatore ed inserita manualmente); 3) valore del coefficiente di temperatura (noto a priori o determinabile al momento), con l’assunzione che l’intervallo tra T attuale e T riferimento sia sufficientemente piccolo da poter stimare lineare l’incremento della conduttività con la temperatura.")
27
Calcolo di una costante di cella
Con la cella di cui voglio determinare la costante misuro col conduttimetro, in una soluzione di KCl 0.1 m, a 25.3°C, una conduttanza di mS. Quanto vale la costante di cella?

28
Acque di acquedotto Acque minerali
A21. Applicazione di misura diretta: valutazione semiquantitativa della durezza(I) Acque di acquedotto Acque minerali La “durezza” è uno dei parametri fondamentali di un’acqua naturale Il termine deriva dalla terminologia delle lavandaie di un tempo, per le quali un’acqua era tanto più “dura” quanto maggiore era la quantità di sapone richiesta per produrre la schiuma. Essa è associata alla presenza di cationi polivalenti in grado di causare la precipitazione di composti insolubili dai saponi alcalini usati come detergenti Il termine “durezza delle acque” deriva dalla terminologia delle lavandaie di un tempo, per le quali un’acqua era tanto più “dura” quanto maggiore era la quantità di sapone richiesta per produrre la schiuma. Questa caratteristica è associata alla presenza di cationi polivalenti in grado di causare la precipitazione di composti insolubili dai saponi alcalini usati come detergenti (motivo per cui ancor oggi nelle macchine per lavaggio vengono impiegati sistemi di "addolcimento"). Si tratta prevalentemente degli ioni calcio e magnesio, benché contribuiscano anche gli altri cationi del secondo gruppo bario e stronzio (e anche altri cationi come ferro, manganese, zinco[1]) [1] Fonte: “Hardness in Drinking-water. Background document for development of WHO Guidelines for Drinking-water Quality”, da Guidelines for drinking-water quality, 2nd ed. Vol. 2. Health criteria and other supporting information. World Health Organization, Geneva, 1996. Si tratta prevalentemente degli ioni Ca2+ e Mg2+, benché contribuiscano anche gli altri cationi del secondo gruppo Ba2+ e Sr2+ (e anche altri cationi come ferro, manganese, zinco…) Si misura in gradi francesi °f definiti come tutti gli ioni di durezza espressi come mg CaCO3 per 100 cm3 di prelievo (oppure gradi francesi °d definiti come tutti gli ioni di durezza espressi come mg CaO per 100 cm3 di prelievo
 Acque di acquedotto. Acque minerali. La durezza è uno dei parametri fondamentali di un’acqua naturale. Il termine deriva dalla terminologia delle lavandaie di un tempo, per le quali un’acqua era tanto più dura quanto maggiore era la quantità di sapone richiesta per produrre la schiuma. Essa è associata alla presenza di cationi polivalenti in grado di causare la precipitazione di composti insolubili dai saponi alcalini usati come detergenti. Il termine durezza delle acque deriva dalla terminologia delle lavandaie di un tempo, per le quali un’acqua era tanto più dura quanto maggiore era la quantità di sapone richiesta per produrre la schiuma. Questa caratteristica è associata alla presenza di cationi polivalenti in grado di causare la precipitazione di composti insolubili dai saponi alcalini usati come detergenti (motivo per cui ancor oggi nelle macchine per lavaggio vengono impiegati sistemi di addolcimento ). Si tratta prevalentemente degli ioni calcio e magnesio, benché contribuiscano anche gli altri cationi del secondo gruppo bario e stronzio (e anche altri cationi come ferro, manganese, zinco[1]) [1] Fonte: Hardness in Drinking-water. Background document for development of WHO Guidelines for Drinking-water Quality , da Guidelines for drinking-water quality, 2nd ed. Vol. 2. Health criteria and other supporting information. World Health Organization, Geneva, Si tratta prevalentemente degli ioni Ca2+ e Mg2+, benché contribuiscano anche gli altri cationi del secondo gruppo Ba2+ e Sr2+ (e anche altri cationi come ferro, manganese, zinco…) Si misura in gradi francesi °f definiti come. tutti gli ioni di durezza espressi come mg CaCO3 per 100 cm3 di prelievo. (oppure gradi francesi °d definiti come. tutti gli ioni di durezza espressi come mg CaO per 100 cm3 di prelievo.")
29
durezza totale = durezza permanente
A22. Applicazione di misura diretta: valutazione semiquantitativa della durezza (II) ebollizione Porzione che scompare se si fa bollire l’acqua. Corrisponde ai bicarbonati dei cationi di metalli alcalino terrosi (che durante l’operazione suddetta precipitano sotto forma dei corrispondenti carbonati con perdita di CO2): ad es. Ca(HCO3)2 CaCO3 + CO2+ H2O Durezza temporanea Cationi di metalli alcalino terrosi (soprattutto Ca2+ e Mg2+) presenti in combinazione con anioni sia di acidi forti (Cl, SO4=, NO3) sia di acidi deboli (HCO3). Durezza totale Durezza permanente Porzione che rimane se si fa bollire l’acqua. Corrisponde ai cationi di metalli alcalino terrosi presenti nel campione in combinazione con anioni di acidi forti durezza totale = durezza permanente + durezza temporanea
 ebollizione. Porzione che scompare se si fa bollire l’acqua. Corrisponde ai bicarbonati dei cationi di metalli alcalino terrosi (che durante l’operazione suddetta precipitano sotto forma dei corrispondenti carbonati con perdita di CO2): ad es. Ca(HCO3)2 CaCO3 + CO2+ H2O. Durezza temporanea. Cationi di metalli alcalino terrosi (soprattutto Ca2+ e Mg2+) presenti in combinazione con anioni sia di acidi forti (Cl, SO4=, NO3) sia di acidi deboli (HCO3). Durezza totale. Durezza permanente. Porzione che rimane se si fa bollire l’acqua. Corrisponde ai cationi di metalli alcalino terrosi presenti nel campione in combinazione con anioni di acidi forti. durezza totale = durezza permanente. + durezza temporanea.")
30
A22. Applicazione di misura diretta:
valutazione semiquantitativa della durezza (III) La titolazione di HCO3- ci fornisce la loro concentrazione molare (mol/dm3). Per trasformarla in °F si procede così: Durezza temporanea [°F] = cHCO3- [mol/dm3] / 2 [mol HCO3- / mol Ca2+ o Mg2+] / 10 [aliquote da 100g soluzione/ 1 dm3] · 100 [g CaCO3 / mol] · 1000 [mg CaCO3 / g CaCO3] La titolazione di Ca2+ + Mg2+ ci fornisce la loro concentrazione molare (mol/dm3). Per trasformarla in °F si procede così: Durezza totale [°F] = (cCa2+ + cMg2+) [mol/dm3] / 10 [aliquote da 100g soluzione/1 dm3] · 100 [g CaCO3/mol] · 1000 [mg CaCO3/g CaCO3] Alcune tra le molte classificazioni: leggere o dolci: durezza inferiore a 15 °F mediamente dure: durezza tra 15 e 30 °F dure: durezza superiore a 30 °F “Calciche” se Ca2+ > 150 mg/dm3 (cioè se Ca2+ da solo è >37.5 °F) Attive su stomaco e fegato; indicate nella gravidanza, nella crescita e nella prevenzione della osteoporosi “Magnesiache” se Mg 2+ > 50 mg/dm3 (cioè se Mg2+ da solo è >20.6 °F) Svolgono prevalentemente attività purgativa; trovano indicazioni anche della prevenzione della arteriosclerosi “Contenenti bicarbonato” se HCO3- > 600 g/dm3 Indicate per stimolare la secrezione gastrica e nelle patologie renali
 La titolazione di HCO3- ci fornisce la loro concentrazione molare (mol/dm3). Per trasformarla in °F si procede così: Durezza temporanea [°F] = cHCO3- [mol/dm3] / 2 [mol HCO3- / mol Ca2+ o Mg2+] / 10 [aliquote da 100g soluzione/ 1 dm3] · 100 [g CaCO3 / mol] · 1000 [mg CaCO3 / g CaCO3] La titolazione di Ca2+ + Mg2+ ci fornisce la loro concentrazione molare (mol/dm3). Per trasformarla in °F si procede così: Durezza totale [°F] = (cCa2+ + cMg2+) [mol/dm3] / 10 [aliquote da 100g soluzione/1 dm3] · 100 [g CaCO3/mol] · 1000 [mg CaCO3/g CaCO3] Alcune tra le molte classificazioni: leggere o dolci: durezza inferiore a 15 °F. mediamente dure: durezza tra 15 e 30 °F. dure: durezza superiore a 30 °F. Calciche se Ca2+ > 150 mg/dm3 (cioè se Ca2+ da solo è >37.5 °F) Attive su stomaco e fegato; indicate nella gravidanza, nella crescita e nella prevenzione della osteoporosi. Magnesiache se Mg 2+ > 50 mg/dm3 (cioè se Mg2+ da solo è >20.6 °F) Svolgono prevalentemente attività purgativa; trovano indicazioni anche della prevenzione della arteriosclerosi. Contenenti bicarbonato se HCO3- > 600 g/dm3. Indicate per stimolare la secrezione gastrica e nelle patologie renali.")
31
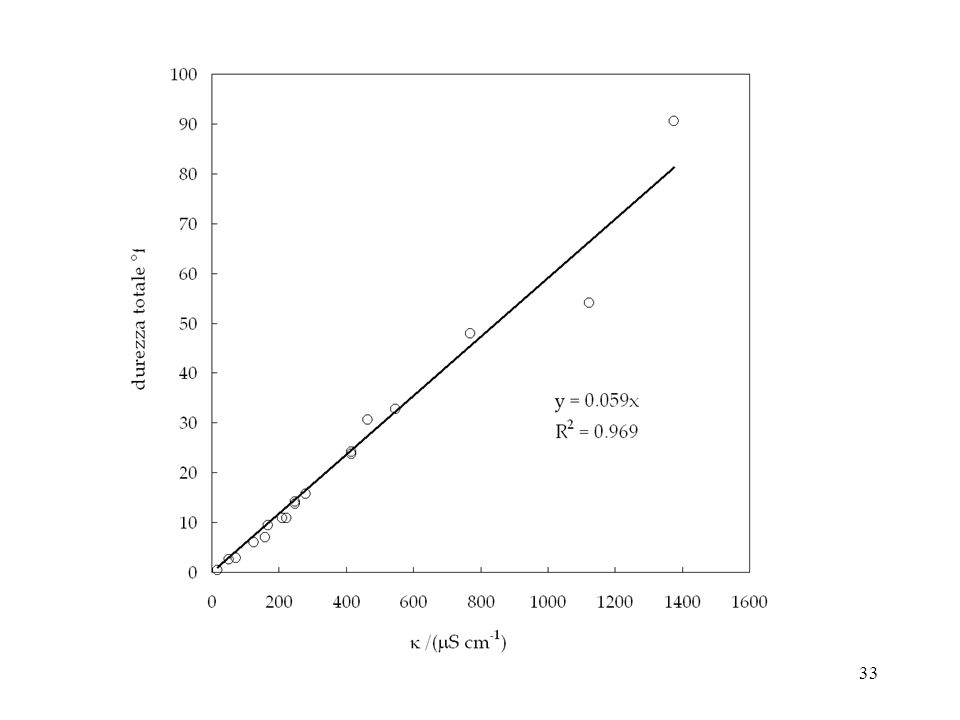
A22. Applicazione di misura diretta:
valutazione semiquantitativa della durezza (IV) Per determinare con precisione (a) la durezza totale e (b) quella temporanea si esegue una titolazione rispettivamente (a) degli ioni calcio e magnesio e (b) degli ioni bicarbonato Per differenza poi si determina la durezza temporanea Tuttavia, la durezza totale delle acque si può stimare con buona approssimazione mediante una semplice misura conduttimetrica. E’ quello che si fa correntemente soprattutto come come prova preliminare per calibrare opportunamente le analisi specifiche successive.
 Per determinare con precisione (a) la durezza totale e (b) quella temporanea. si esegue una titolazione rispettivamente. (a) degli ioni calcio e magnesio e (b) degli ioni bicarbonato. Per differenza poi si determina la durezza temporanea. Tuttavia, la durezza totale delle acque si può stimare con buona approssimazione. mediante una semplice misura conduttimetrica. E’ quello che si fa correntemente soprattutto come come prova preliminare. per calibrare opportunamente le analisi specifiche successive.")
32
Alcune acque minerali da tavola italiane

34
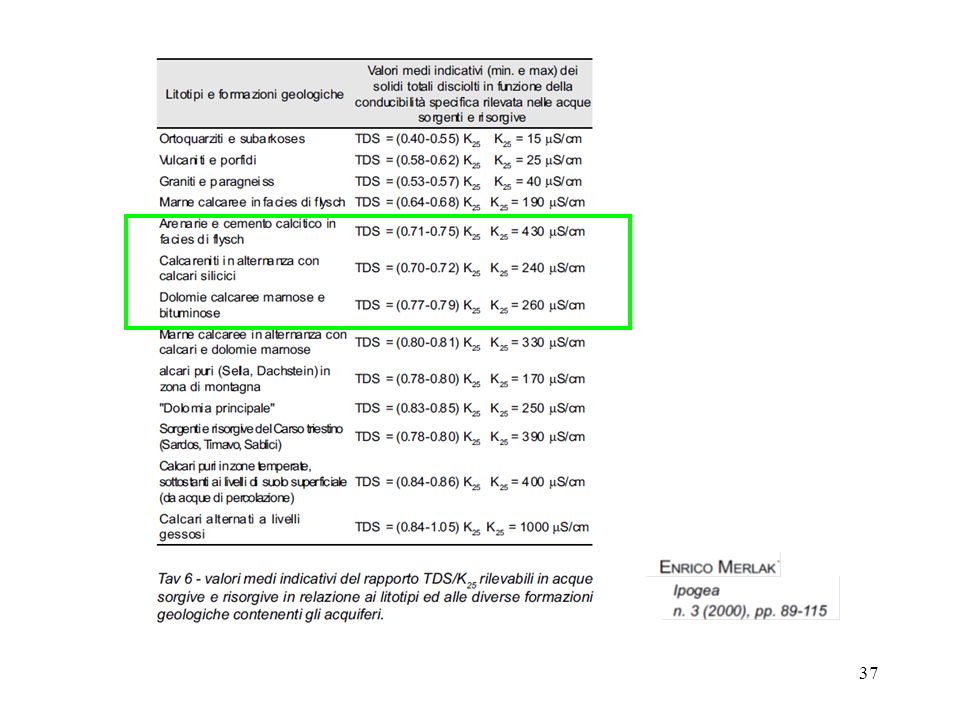
A23. Applicazione di misura diretta:
valutazione semiquantitativa del residuo fisso o solidi totali disciolti Il residuo fisso è un parametro utilizzato per classificare le acque minerali e le acque potabili in generale. Solitamente espresso in ppm (mg/dm3), indica la quantità di sostanza solida perfettamente secca che rimane dopo aver fatto evaporare in una capsula di platino, previamente tarata, una quantità nota di acqua precedentemente filtrata. Essiccare la capsula preliminarmente in stufa per circa 1 ora, alla temperatura di 180 ± 2°C, fino a peso costante. Filtrare il campione su di un filtro a 0,45 μm. Prelevare un’aliquota del campione di acqua filtrata che possa presumibilmente fornire un residuo compreso tra 25 e 250 mg e preferibilmente tra 100 e 250 mg. Un calcolo preliminare fatto in base al valore della conduttività è normalmente sufficiente per determinare il volume da evaporare. Porre l'aliquota misurata del campione d'acqua nella capsula tarata ed evaporare sino a piccolo volume con lampada a quarzo o bagno a sabbia, evitando l'ebollizione. Completare l'evaporazione dell'acqua trasferendo la capsula in stufa ed innalzando progressivamente la temperatura fino a 180 °C. Essiccare fino a peso costante (si considera peso costante quello ottenuto quando la variazione di peso riscontrata in due cicli successivi di riscaldamento, raffreddamento e pesata non superi 0.5 mg). (eliminando così i sali di ammonio più volatili ed alcune sostanze organiche) Pesare la capsula subito dopo averla fatta raffreddare in essiccatore. Si può poi riscaldarla ulteriormente a 500 °C distruggendo tutti i sali di ammonio, le sostanze organiche ed i nitrati. Il risultato si esprime in ppm (parti per milione) oppure in mg/L, specificando sempre a quale temperatura ci si riferisce (residuo fisso a 180 °C o residuo fisso a 500 °C). acque meteoriche: compreso tra 10 e 80 mg/L acque oligominerali: compreso tra 80 e 200 mg/L acque mediominerali: compreso tra 200 e mg/L acque minerali: superiore a mg/L acque salate: superiore a mg/L.
, indica la quantità di sostanza solida perfettamente secca che rimane dopo aver fatto evaporare in una capsula di platino, previamente tarata, una quantità nota di acqua precedentemente filtrata. Essiccare la capsula preliminarmente in stufa per circa 1 ora, alla temperatura di 180 ± 2°C, fino a peso costante. Filtrare il campione su di un filtro a 0,45 μm. Prelevare un’aliquota del campione di acqua filtrata che possa presumibilmente fornire un residuo. compreso tra 25 e 250 mg e preferibilmente tra 100 e 250 mg. Un calcolo preliminare fatto in base al. valore della conduttività è normalmente sufficiente per determinare il volume da evaporare. Porre l aliquota misurata del campione d acqua nella capsula tarata ed evaporare sino a piccolo volume con lampada a quarzo o bagno a sabbia, evitando l ebollizione. Completare l evaporazione dell acqua trasferendo la capsula in stufa ed innalzando progressivamente la temperatura fino a 180 °C. Essiccare fino a peso costante (si considera peso costante quello ottenuto quando la variazione di peso riscontrata in due cicli successivi di riscaldamento, raffreddamento e pesata non superi 0.5 mg). (eliminando così i sali di ammonio più volatili ed alcune sostanze organiche) Pesare la capsula subito dopo averla fatta raffreddare in essiccatore. Si può poi riscaldarla ulteriormente a 500 °C distruggendo tutti i sali di ammonio, le sostanze organiche ed i nitrati. Il risultato si esprime in ppm (parti per milione) oppure in mg/L, specificando sempre a quale temperatura ci si riferisce (residuo fisso a 180 °C o residuo fisso a 500 °C). acque meteoriche: compreso tra 10 e 80 mg/L. acque oligominerali: compreso tra 80 e 200 mg/L. acque mediominerali: compreso tra 200 e mg/L. acque minerali: superiore a mg/L. acque salate: superiore a mg/L.")
35
Alcune acque minerali da tavola italiane

36
Residuo fisso

38
A24. Applicazione di misura diretta:
monitoraggio dell’acidità libera in acidi organici ad elevatissima concentrazione Problema: stimare l’acidità di soluzioni per l’esfoliazione della pelle, prima di sperimentarle su pazienti Effettuando misure di pH con il protocollo acquoso (standard, elettrodo di riferimento, ponte salino) si osserva che risultano raggruppate in un range molto ristretto (1.5 unità pH), molto poco differenziate a seconda dell’acido, e soprattutto non seguono affatto la formulazione del mezzo acquo-organico In questo importante caso applicativo il pH risulta inutilizzabile per scopi di formulazione e controllo
 si osserva che risultano raggruppate in un range molto ristretto (1.5 unità pH), molto poco differenziate a seconda dell’acido, e soprattutto non seguono affatto la formulazione del mezzo acquo-organico. In questo importante caso applicativo il pH risulta inutilizzabile per scopi di formulazione e controllo.")
39
Una possibile soluzione del problema: monitorare l’acidità libera attraverso la conduttività
La conduttività è una grandezza intrinsecamente non selettiva ma “integrale” perché dipende dai prodotti (carica × concentrazione × mobilità) di tutti gli ioni liberi nella soluzione studiata Carica ione Concentrazione ione Mobilità ione Tuttavia, a) in soluzioni acquose o miste ma con percentuale significativa di acqua il contributo di gran lunga più significativo alla conduttività é quello degli ioni H+ b) nel nostro caso paragoniamo soluzioni di acidi deboli monoprotici HA a solvente costante oppure lo stesso acido al variare del solvente o della temperatura la grandezza si può considerare un sensibile e affidabile indicatore di disponibilità e mobilità dei protoni
 di tutti gli ioni liberi nella soluzione studiata. Carica ione. Concentrazione. ione. Mobilità ione. Tuttavia, a) in soluzioni acquose o miste ma con percentuale significativa di acqua il contributo di gran lunga più significativo alla conduttività é quello degli ioni H+ b) nel nostro caso paragoniamo soluzioni di acidi deboli monoprotici HA a solvente costante oppure lo stesso acido al variare del solvente o della temperatura la grandezza si può considerare un sensibile e affidabile indicatore di disponibilità e mobilità dei protoni.")
40
Costanti di dissociazione acida Ka nettamente e correttamente differenziate
Andamenti lievemente parabolici estremamente regolari La conduttività cala regolarmente al calare della % DMI Il calo è sempre più accentuato all’aumentare della forza dell’acido

41
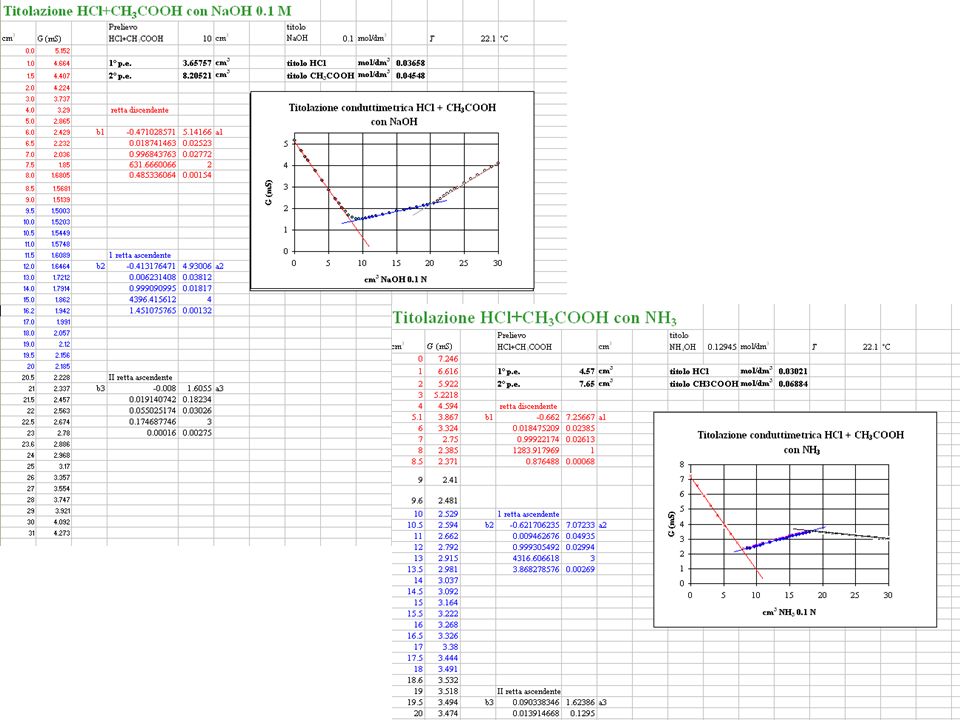
A25. Applicazione come parametro per seguire titolazioni:
titolazioni conduttimetriche acido/base (I) In generale nelle titolazioni per neutralizzazione, precipitazione, scambio, ecc. ci si possono attendere cambiamenti di conduttività che si possono sfruttare per seguire il decorso della reazione e soprattutto individuare il punto finale. Ovviamente si può eseguire una titolazione conduttimetrica seguendo della soluzione la conduttività o anche solo la conduttanza, che è ad essa proporzionale. La temperatura deve essere pressochè costante, ed è preferibile che anche il volume della soluzione non cambi troppo durante la titolazione. A differenza delle curve di titolazione potenziometriche, i punti di equivalenza non corrispondono a flessi, ma ad angoli d’intersezione tra tratti rettilinei. L’accuratezza del metodo è tanto maggiore quanto più acuto è l’angolo d’intersezione, e quanto maggiore è la correlazione lineare dei punti sperimentali; non hanno particolare senso accurate misure vicino al punto di equivalenza, anzi, a causa di idrolisi, dissociazione, o solubilità del prodotto della reazione nei pressi del punto di equivalenza il grafico di titolazione in tale zona risulta arrotondato e quindi il punto di equivalenza si valuta estrapolando i tratti rettilinei e valutandone il punto d’intersezione. I vantaggi delle titolazioni conduttimetriche rispetto a quelle colorimetriche e potenziometriche sono: Il metodo è accurato tanto in soluzioni diluite che concentrate Il metodo funziona bene in casi altrimenti critici come titolazioni di acidi deboli con basi deboli e titolazioni di acidi debolissimi come fenoli, acido borico... Rispetto alle colorimetriche: Il metodo funziona anche con soluzioni torbide
 In generale nelle titolazioni per neutralizzazione, precipitazione, scambio, ecc. ci si possono attendere cambiamenti di conduttività che si possono sfruttare per seguire il decorso della reazione e soprattutto individuare il punto finale. Ovviamente si può eseguire una titolazione conduttimetrica seguendo della soluzione la conduttività o anche solo la conduttanza, che è ad essa proporzionale. La temperatura deve essere pressochè costante, ed è preferibile che anche il volume della soluzione non cambi troppo durante la titolazione. A differenza delle curve di titolazione potenziometriche, i punti di equivalenza non corrispondono a flessi, ma ad angoli d’intersezione tra tratti rettilinei. L’accuratezza del metodo è tanto maggiore quanto più acuto è l’angolo d’intersezione, e quanto maggiore è la correlazione lineare dei punti sperimentali; non hanno particolare senso accurate misure vicino al punto di equivalenza, anzi, a causa di idrolisi, dissociazione, o solubilità del prodotto della reazione nei pressi del punto di equivalenza il grafico di titolazione in tale zona risulta arrotondato e quindi il punto di equivalenza si valuta estrapolando i tratti rettilinei e valutandone il punto d’intersezione. I vantaggi delle titolazioni conduttimetriche rispetto a quelle colorimetriche e potenziometriche sono: Il metodo è accurato tanto in soluzioni diluite che concentrate. Il metodo funziona bene in casi altrimenti critici come titolazioni di acidi deboli con basi deboli e titolazioni di acidi debolissimi come fenoli, acido borico... Rispetto alle colorimetriche: Il metodo funziona anche con soluzioni torbide.")
42
cala forza o concentrazione acido
A24. Applicazione come parametro per seguire titolazioni: titolazioni conduttimetriche acido/base (II) (o G) cala forza o concentrazione acido cala forza base VT Hp: temperatura, volume e mobilità ioniche circa costanti
 (o G) cala forza o concentrazione acido. cala forza. base. VT. Hp: temperatura, volume e mobilità ioniche circa costanti.")
43
Nel primo tratto la conduttività cala perchè lo ione H+ (il più mobile, °H+ 350 1cm2mol1) viene sostituito da Me+ (°Me+ 40/80 1cm2 mol1); dopo il punto di equivalenza ricresce al crescere dell’eccesso di OH (°OH 198 1cm2 mol1).
 viene sostituito da Me+ (°Me+ 40/80 1cm2 mol1); dopo il punto di equivalenza ricresce al crescere dell’eccesso di OH (°OH 198 1cm2 mol1).")
44
Nel primo tratto la conduttività cala perchè lo ione H+ (il più mobile, °H+ 350 1cm2mol1) viene sostituito da NH4+; dopo il punto di equivalenza resta pressochè costante perchè NH3 in eccesso ha su di essa un’influenza trascurabile rispetto al sale (NH4)2SO4 .
 viene sostituito da NH4+; dopo il punto di equivalenza resta pressochè costante perchè NH3 in eccesso ha su di essa un’influenza trascurabile rispetto al sale (NH4)2SO4 .")
45
Il primo tratto arrotondato è dato dal fatto che G tende a calare perchè AcONa formatosi reprime la ionizzazione di AcOH ancora presente, ma presto ricresce al crescere della forza ionica del sale.

46
In questo caso la curva è più facilmente utilizzabile.

48
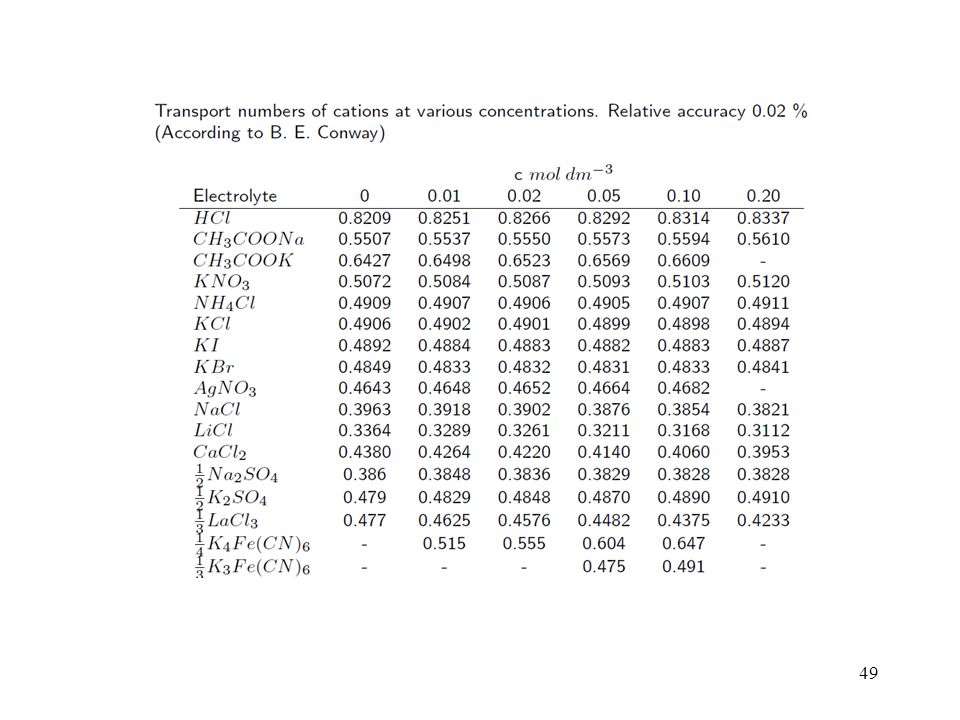
Membrana permeoselettiva:
A26. Grandezze relative che esprimono il trasporto elettrico in soluzione numero di trasporto ti = Naturalmente, ti = 1. numeri di trasporto “segnati” o di Scatchard i i = moli di specie i che attraversano una giunzione di riferimento all'interno di una catena galvanica nel verso di movimento dei cationi (in una pila, dal polo – al polo +) per il passaggio di carica di 1 Faraday. Questa definizione è vantaggiosa perché permette di attribuire un numero di trasporto anche a specie non cariche come il solvente, i numeri di trasporto hanno un segno che indica il verso di movimento delle specie corrispondenti. In particolare, per le specie cariche con carica zi risulta ti = ti/zi (con la carica presa col suo segno) , e quindi anche ti zi = 1. extratermodinamiche dEL = kidlogai + kSdlogaS Potenziale interliquido: Su tutti gli ioni presenti 1 solvente a diversa attività, o 2 solventi diversi alla giunzione (caso particolare) dEM = k(idlogai + jdlogaj) kidlogai k(ti/zi)dlogai EM (k/zi)log(ai”/ai’) Potenziale di membrana: Membrana permeoselettiva: i 1 (ione primario) j 0 (ioni interferenti)
 per il passaggio di carica di 1 Faraday. Questa definizione è vantaggiosa perché. permette di attribuire un numero di trasporto anche a specie non cariche come il solvente, i numeri di trasporto hanno un segno che indica il verso di movimento delle specie corrispondenti. In particolare, per le specie cariche con carica zi risulta ti = ti/zi (con la carica presa col suo segno) , e quindi anche ti zi = 1. extratermodinamiche. dEL = kidlogai + kSdlogaS. Potenziale interliquido: Su tutti gli ioni presenti. 1 solvente a diversa attività, o 2 solventi diversi alla giunzione. (caso particolare) dEM = k(idlogai + jdlogaj) kidlogai k(ti/zi)dlogai EM (k/zi)log(ai /ai’) Potenziale di membrana: Membrana permeoselettiva: i 1 (ione primario) j 0 (ioni interferenti)")
50
Variazione delle conduttività molari limite di alcuni ioni con la temperatura
Ritorno

Presentazioni simili











1 VELOCITA DI REAZIONE ED EQUILIBRI.>")
>")
1 Soluzioni e sospensioni.>")





