Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
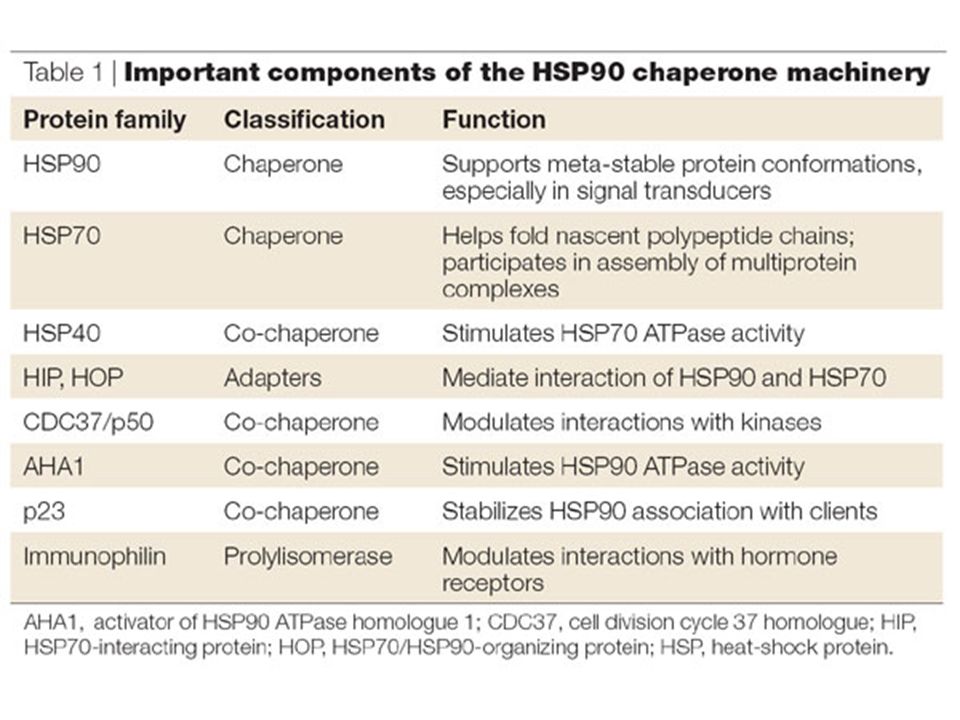
HSP90 (Heat-Shock Protein 90 kDa)
")
2
Participation of molecular chaperones in regulating many aspects of posttranslational
protein homeostasis. Newly synthesized, conformationally labile client proteins associate with multi-protein complexes that contain various chaperones, co-chaperones and accessory molecules (different coloured shapes). The particular components of a complex vary according to the client and also help specify the function of a particular complex. Dynamic association of a client with chaperone complexes can prevent its aggregation (a) and assist in its intracellular trafficking, especially its translocation across membranous structures such as the endoplasmic reticulum (ER) (b). For many clients involved in signal transduction pathways, association with the chaperone machinery maintains the protein in a meta-stable state that allows it to be activated by specific stimuli such as ligand binding, phosphorylation or assembly into multisubunit signalling complexes (c). In the absence of appropriate stimuli, chaperone complexes can target the client for degradation through the ubiquitin–proteasome pathway, thereby regulating its steady-state cellular level (d).
. The particular. components of a complex vary according to the client and also help specify the function of a. particular complex. Dynamic association of a client with chaperone complexes can prevent its. aggregation (a) and assist in its intracellular trafficking, especially its translocation across. membranous structures such as the endoplasmic reticulum (ER) (b). For many clients involved. in signal transduction pathways, association with the chaperone machinery maintains the. protein in a meta-stable state that allows it to be activated by specific stimuli such as ligand. binding, phosphorylation or assembly into multisubunit signalling complexes (c). In the. absence of appropriate stimuli, chaperone complexes can target the client for degradation. through the ubiquitin–proteasome pathway, thereby regulating its steady-state cellular level (d).")
3
The numbering 1–732 indicates the approximate positions in the amino acid sequence of the human protein that define its functional domains. 'CR' refers to a charged region which serves as a flexible linker between the N-terminal and middle domains. The locations where various small molecules bind HSP90 (heat-shock protein of 90 kDa) and modulate its function are indicated. The biochemical functions of each domain are also shown. 17AAG, 17-allylaminogeldanamycin; GA, geldanamycin.
 and modulate its function are indicated. The biochemical functions of each domain are also shown. 17AAG, 17-allylaminogeldanamycin; GA, geldanamycin..")
4
Model for the altered states of Hsp90
Model for the altered states of Hsp90. The native uncomplexed form predominates in normal cells and tissues and has low ATPase activity and low 17AAG affinity. Conversion of the native form to the super-chaperone complex is driven by the accumulation of overexpressed and mutated client proteins in cancer cells, together with tumour microenvironmental stresses such as hypoxia, nutrient deprivation and the build-up of metabolic products. The heightened sensitivity of the super-chaperone-complex form of Hsp90 in cancer cells provides an explanation for the therapeutic selectivity of the drug, which now shows promise in early clinical trials. In the paper by Kamal et al.[ 8 ], the more sensitive super-chaperone complex in cancer cells is shown to contain Hsp40, p23, Hop and Hsp70. It is probable that the super-chaperone complex also contains other proteins, including Aha1 and Cdc37.

6
Model for tumor selectivity of Hsp90 inhibitors and Hsp90-dependent malignant progression. Hsp90 in normal cells exists in an uncomplexed form that has low-affinity for Hsp90 inhibitor drugs, which accumulate poorly in normal tissues, and normal cells exhibit poor drug sensitivity. By contrast, the Hsp90 in cancer cells is involved in the active chaperoning of overexpressed oncoproteins and exists in a complexed form with co-chaperone proteins (p23 and Hop are not in the same complex). Complexed Hsp90 in cancer cells exhibits high-affinity binding to Hsp90 inhibitor drugs, which accumulate in tumor tissues, and tumor cells exhibit good drug sensitivity. This model predicts that the accumulation of mutant proteins in advanced cancer would further increase Hsp90 usage and make tumor cells more Hsp90 dependent. Furthermore, this model suggests that the high-affinity change of Hsp90 can be driven by the overexpression of oncoproteins, as well as by stressful conditions in normal cells (e.g. heat).
. Complexed Hsp90 in cancer cells exhibits high-affinity binding to Hsp90 inhibitor drugs, which accumulate in tumor tissues, and tumor cells exhibit good drug sensitivity. This model predicts that the accumulation of mutant proteins in advanced cancer would further increase Hsp90 usage and make tumor cells more Hsp90 dependent. Furthermore, this model suggests that the high-affinity change of Hsp90 can be driven by the overexpression of oncoproteins, as well as by stressful conditions in normal cells (e.g. heat)..")
9
The numbering 1–732 indicates the approximate positions in the amino acid sequence of the human protein that define its functional domains. 'CR' refers to a charged region which serves as a flexible linker between the N-terminal and middle domains. The locations where various small molecules bind HSP90 (heat-shock protein of 90 kDa) and modulate its function are indicated. The biochemical functions of each domain are also shown. 17AAG, 17-allylaminogeldanamycin; GA, geldanamycin.
 and modulate its function are indicated. The biochemical functions of each domain are also shown. 17AAG, 17-allylaminogeldanamycin; GA, geldanamycin..")
10
Figure 3 Schematic of the PI3K–AKT/PKB and RAS–RAF–MEK–ERK1/2 pathways demonstrating the components which are reliant on HSP90 chaperone function. 90, HSP90; P, phosphate.

11
a | The newly synthesized oestrogen receptor (ER) associates with HSP70 (heat-shock protein of 70 kDa), HSP40 and the adapter HIP (HSP70-interacting protein) to form an early complex. The hydrophobic hormone-binding domain is partially exposed in this complex and HSP90 binds to this region in association with the adapter protein HOP (HSP70/HSP90-organizing protein) and displaces HSP40 to form an intermediate complex. In an ATP-dependent manner, HSP90 fully exposes the hormone-binding domain, and the co-chaperone p23 stabilizes the ATP-bound HSP90. Cyclophilin 40 (CYP40) fills the open tetratricopeptide repeat (TPR) acceptor site on HSP90 to complete a mature complex. In the absence of oestrogenic ligands, the ER is released from the mature complex to undergo additional cycles of chaperone interactions. Oestrogen binding, however, leads to a conformational change in the ER, which releases chaperone components and leads to tight binding of the receptor protein to oestrogen response elements and the recruitment of the co-activators needed to drive transcription.
 and displaces HSP40 to form an intermediate complex. In an ATP-dependent manner, HSP90 fully exposes the hormone-binding domain, and the co-chaperone p23 stabilizes the ATP-bound HSP90. Cyclophilin 40 (CYP40) fills the open tetratricopeptide repeat (TPR) acceptor site on HSP90 to complete a mature complex. In the absence of oestrogenic ligands, the ER is released from the mature complex to undergo additional cycles of chaperone interactions. Oestrogen binding, however, leads to a conformational change in the ER, which releases chaperone components and leads to tight binding of the receptor protein to oestrogen response elements and the recruitment of the co-activators needed to drive transcription..")
12
b | Geldanamycin (GA) binding to HSP90 locks the chaperone in an alternative conformation that prevents normal cycling and the formation of mature chaperone complexes. The ER accumulates in an intermediate complex that recruits E3 ubiquitin ligase and drives proteasome-mediated degradation of the protein, thereby dramatically lowering cellular levels of the receptor and disrupting its function.

13
a | Wild-type p53 is held in dynamic equilibrium by transient association with HSP90 (heat-shock protein of 90 kDa)-containing complexes (presumably containing p23 and CYP40 (cyclophilin 40)) that maintain it in a conformation that can be activated for DNA binding. In the absence of DNA damage, HSP90 (presumably in conjunction with its usual co-chaperones HIP (HSP70-interacting protein), HOP (HSP70/HSP90-organizing protein) and HSP70) presents p53 for degradation by recruiting ubiquitin ligases, such as MDM2 and CHIP (carboxy-terminus of HSP70-interacting protein), to the protein and stimulating its proteasome-mediated degradation. This maintains the low, steady-state level of wild-type p53 in normal cells.
, HOP (HSP70/HSP90-organizing protein) and HSP70) presents p53 for degradation by recruiting ubiquitin ligases, such as MDM2 and CHIP (carboxy-terminus of HSP70-interacting protein), to the protein and stimulating its proteasome-mediated degradation. This maintains the low, steady-state level of wild-type p53 in normal cells..")
14
b | Most mutant p53 proteins are not able to achieve a conformation that is capable of DNA binding despite extended chaperone interactions. Presentation of p53 for ubiquitylation by MDM2, and possibly CHIP, is also impaired, which leads to the accumulation of aggregation-prone, dysfunctional protein. The binding of geldanamycin (GA) to HSP90 inhibits normal chaperone cycling and drives the degradation of p53 mutants, which leads to a decrease in their cellular levels. It remains unclear whether this decrease is sufficient to disrupt the dominant-negative and positive tumour-promoting activities of such mutants.
 to HSP90 inhibits normal chaperone cycling and drives the degradation of p53 mutants, which leads to a decrease in their cellular levels. It remains unclear whether this decrease is sufficient to disrupt the dominant-negative and positive tumour-promoting activities of such mutants..")
15
Proteasoma

16
In addition to ubiquitylation, eukaryotic cells have evolved additional signalling systems utilizing other polypeptides that share the same fold as ubiquitin. Examples of such ubiquitin-like proteins (Ubls)169 are shown in the table and include NEDD8, SUMO and ISG15. The Ubls are conjugated to their target proteins in a manner that mechanistically resembles ubiquitylation but have dramatically different functional consequences. Instead of promoting proteasomal degradation, Ubl conjugation modifies target activity or subcellular localization. Moreover, conjugation of a single ubiquitin molecule (that is, monoubiquitylation) — by contrast with polyubiquitylation — has been shown to regulate target activity instead of enforcing its destruction. Several classes of ubiquitin-binding proteins have been identified, and many of these interact with mono-ubiquitylated forms of ubiquitin (for example, Cue domain containing proteins). Thus, mono-ubiquitylation and Ubl conjugation can be conceptualized as a canonical post-translational modification that employs proteins (ubiquitin or Ubl) instead of a small functional group such as phosphate or acetyl groups. In addition to K48-linked polyubiquitin chains, other classes of conjugation products, such as K64-linked chains and branched polyubiquitin chains, can also be formed on targets. To date, K63-chains have been implicated in controlling protein activity and appear to have no role in protein degradation. Type II Ubls contain other functional domains in addition to the ubiquitin-fold motif. They are generally larger protein molecules that are not conjugated to other proteins. Instead, they perform multiple and incompletely understood cellular functions that might or might not intersect with the ubiquitin–proteasome pathway of protein degradation, such as DNA repair, cell cycle control or serving as E3 ligases. One of the type II ubiquitin-like proteins, parkin, has been implicated in familial Parkinson's disease as a putative ubiquitin ligase.
 — by contrast with polyubiquitylation — has been shown to regulate target activity instead of enforcing its destruction. Several classes of ubiquitin-binding proteins have been identified, and many of these interact with mono-ubiquitylated forms of ubiquitin (for example, Cue domain containing proteins). Thus, mono-ubiquitylation and Ubl conjugation can be conceptualized as a canonical post-translational modification that employs proteins (ubiquitin or Ubl) instead of a small functional group such as phosphate or acetyl groups. In addition to K48-linked polyubiquitin chains, other classes of conjugation products, such as K64-linked chains and branched polyubiquitin chains, can also be formed on targets. To date, K63-chains have been implicated in controlling protein activity and appear to have no role in protein degradation. Type II Ubls contain other functional domains in addition to the ubiquitin-fold motif. They are generally larger protein molecules that are not conjugated to other proteins. Instead, they perform multiple and incompletely understood cellular functions that might or might not intersect with the ubiquitin–proteasome pathway of protein degradation, such as DNA repair, cell cycle control or serving as E3 ligases. One of the type II ubiquitin-like proteins, parkin, has been implicated in familial Parkinson s disease as a putative ubiquitin ligase.")
17
Protein degradation through the UPS is a highly regulated process, involving several steps3, 38. The first step in the cascade is ubiquitin activation by E1 (ubiquitin-activating enzyme) followed by ubiquitin delivery to E2 (ubiquitin-conjugating enzyme) (a). The second step involves complex formation by E2-Cys Ub, E3 (ubiquitin ligase) and the substrate (b). These initial steps involve formation of thiol esters between the active-site cysteines of E1 and E2 enzymes and the carboxy-terminal carboxylate of ubiquitin. The third step (c) comprises transfer of ubiquitins to the substrate lysine(s) to earmark the substrate with a polyubiquitin chain. In the fourth step of the pathway (d), a polyubiquitylated substrate is released from the E3. Proteasomes recognize the polyubiquitin chain as a signal to de-ubiquitylate and destroy the substrate. The fifth step seals the fate of a doomed protein (e). The proteasome unfolds the substrate in ATP-dependent manner, removes the ubiquitin chain through a proteasome-associated ubiquitin hydrolase activity, and threads the unfolded protein into the proteasome chamber, where the protease active sites are located. The ubiquitin molecules are recycled, and the peptides generated are used in major histocompatibility class I-coupled antigen presentation or degraded to amino acids that are recycled for new protein synthesis. MDM2, double minute 2; SCF, SKP1–Cullin–F-box; SKP, S-phase kinase-associated protein; RBX, RING-box protein.
 followed by ubiquitin delivery to E2 (ubiquitin-conjugating enzyme) (a). The second step involves complex formation by E2-Cys Ub, E3 (ubiquitin ligase) and the substrate (b). These initial steps involve formation of thiol esters between the active-site cysteines of E1 and E2 enzymes and the carboxy-terminal carboxylate of ubiquitin. The third step (c) comprises transfer of ubiquitins to the substrate lysine(s) to earmark the substrate with a polyubiquitin chain. In the fourth step of the pathway (d), a polyubiquitylated substrate is released from the E3. Proteasomes recognize the polyubiquitin chain as a signal to de-ubiquitylate and destroy the substrate. The fifth step seals the fate of a doomed protein (e). The proteasome unfolds the substrate in ATP-dependent manner, removes the ubiquitin chain through a proteasome-associated ubiquitin hydrolase activity, and threads the unfolded protein into the proteasome chamber, where the protease active sites are located. The ubiquitin molecules are recycled, and the peptides generated are used in major histocompatibility class I-coupled antigen presentation or degraded to amino acids that are recycled for new protein synthesis. MDM2, double minute 2; SCF, SKP1–Cullin–F-box; SKP, S-phase kinase-associated protein; RBX, RING-box protein..")
18
Attivazione dell’ubiquitina
adenilazione trasferimento a E2 Coniugazione Riconoscimento della catena di poliubiquitina da parte delle subunità regolatrici del proteasoma Rimozione della catena di poliubiquitina Degradazione enzimatica del substrato

19
This step of the ubiquitylation cascade requires ATP binding to the ATP-binding cleft of E1. This process can be divided into four major sub-steps: (a) adenylation of the carboxyl terminus of a free ubiquitin molecule by E1; (b) rapid cis transfer of the E1-bound ubiquitin molecule from AMP to the active-site cysteine in E1, with subsequent release of free AMP; (c) adenylation of another free ubiquitin residue by the same Cys Ub-loaded E1 molecule; and (d) recruitment of E2 (ubiquitin-conjugating enzyme) followed by transfer of the activated ubiquitin from the active cysteine of E1 to the catalytic cysteine of E2.
 adenylation of the carboxyl terminus of a free ubiquitin molecule by E1; (b) rapid cis transfer of the E1-bound ubiquitin molecule from AMP to the active-site cysteine in E1, with subsequent release of free AMP; (c) adenylation of another free ubiquitin residue by the same Cys Ub-loaded E1 molecule; and (d) recruitment of E2 (ubiquitin-conjugating enzyme) followed by transfer of the activated ubiquitin from the active cysteine of E1 to the catalytic cysteine of E2..")
20
a | The ubiquitin cascade is pyramidal in design
a | The ubiquitin cascade is pyramidal in design. A single E1-activating enzyme transfers ubiquitin to roughly three dozen E2s, which function together with several hundred different E3 ubiquitin ligases to ubiquitylate thousands of substrates.

21
b | Classes of ubiquitin ligases: single RING-finger E3s; HECT E3s; and multi-subunit RING-finger E3s, exemplified by the SCF complexes. Only HECT-domain E3s form a covalent bond with ubiquitin during polyubiquitylation of their target proteins. Specific E3s discussed in this review are indicated.

22
a | Schematic representation of a C3HC4 RING finger
a | Schematic representation of a C3HC4 RING finger. Most RING fingers contain two zinc atoms (yellow) coordinated with cysteine or cysteine/histidine-rich clusters (red). The general consensus sequence is: C-X2-C-X9–39-C-X1–3-H-X2–3-C-X2-C-X4–48-C-X2-C, although some variations exist.
 coordinated with cysteine or cysteine/histidine-rich clusters (red). The general consensus sequence is: C-X2-C-X9–39-C-X1–3-H-X2–3-C-X2-C-X4–48-C-X2-C, although some variations exist.")
23
a | Simplified view of p53 signalling pathway
a | Simplified view of p53 signalling pathway. Genotoxic stress activates a network of protein kinase pathways that converge on the p53 protein. Although unphosphorylated p53 is rapidly degraded via the ubiquitin–proteasome system, phosphorylated p53 cannot bind its primary E3 ligase (MDM2) and is stabilized. Stabilized p53 arrests the cell cycle, and promotes DNA repair and apoptosis, depending on the cellular context. Failure of the p53 response is the most common event in human cancers, and restoration of p53 stabilization is a well-established potential anticancer strategy.
 and is stabilized. Stabilized p53 arrests the cell cycle, and promotes DNA repair and apoptosis, depending on the cellular context. Failure of the p53 response is the most common event in human cancers, and restoration of p53 stabilization is a well-established potential anticancer strategy.")
24
. b | Structure of Nutlin (left) and RITA (right), the first small-molecule inhibitors of the p53–MDM2 interaction73, 75. c | Structure of the Nutlin–p53 complex19. Left: the sides of p53-binding groove in MDM2 (red) is limited by two -helices and a short -sheet while the bottom is formed by two shorter -helices perpendicular to the pocket sides. The amino-terminal domain of p53 (green) is stabilized in -helical conformation upon binding to MDM2 due to a network of hydrogen bonds trapping three crucial residues of p53, Phe19, Trp23 and Leu26. These crucial p53 residues point towards the bottom of the groove. Right: Nutlin prevents interaction between p53 and MDM2 by mimicking the conformation of Phe19, Trp23 and Leu26 of p53 to block the MDM2 p53-interacting domain73. MDM2, double minute 2.
 is limited by two -helices and a short -sheet while the bottom is formed by two shorter -helices perpendicular to the pocket sides. The amino-terminal domain of p53 (green) is stabilized in -helical conformation upon binding to MDM2 due to a network of hydrogen bonds trapping three crucial residues of p53, Phe19, Trp23 and Leu26. These crucial p53 residues point towards the bottom of the groove. Right: Nutlin prevents interaction between p53 and MDM2 by mimicking the conformation of Phe19, Trp23 and Leu26 of p53 to block the MDM2 p53-interacting domain73. MDM2, double minute 2..")
25
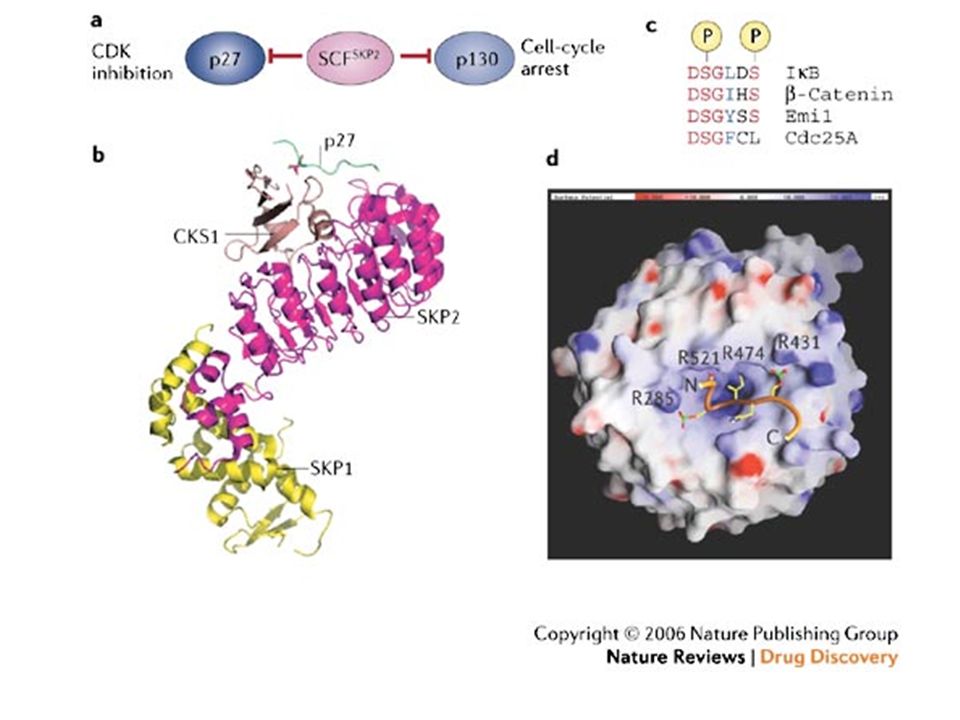
. c | SCF E3 complexes consists of a scaffold-like cullin molecule; a RING-finger-containing subunit (RBX1 or RBX2), which functions to bind E2s; and a substrate-specificity module, which binds substrates. d | Distinct cullins utilize structurally related specificity factors that are specific to each cullin. β-TRCP, β-transducin repeat-containing protein; BTB, broad complex/tracktrack/bric-a-brac; DDB, DNA-damage binding protein; FBW, F-box and WD40 domain protein; HECT, homologous to E6-AP COOH-terminus; RBX, RING-box protein; SCF, SKP1–Cullin–F-box; SKP, S-phase kinase-associated protein; VHL, von Hippel-Lindau.

27
Proteasome structure. A three-dimensional representation of the proteasome multi-enzyme complex. This is composed of the 20S complex, which comprises - and -subunits, and two 19S regulatory complexes. Together with ATP, these form the 26S proteasome.

28
20

30
BORTEZOMIB (VELCADE®)
The effects of proteasome inhibition on the stability Figure 3 The structure of the dipeptidyl boronic acid proteasome inhibitor bortezomib. BORTEZOMIB (VELCADE®)
")
31
Figure 4 Cross-sectional view of the bortezomib binding site
in the proteasome. Bortezomib interacts with a threonine residue located on the b subunit that confers chymotryptic proteolytic activity.

34
Cellular pathways associated with the proteasome
Cellular pathways associated with the proteasome. a | The CDC25 family and the cyclins are intimately involved in cell-cycle regulation and are catabolysed by proteasomes in a tightly controlled sequence. With proteasome inhibition, the regulation of CDC25A, CDC25C, KIP1 and the cyclins is destabilized, which is thought to make the cell more susceptible to apoptosis. b | The tumour suppressor p53 accumulates with cellular stress, such as DNA damage, oncogene activation and hypoxia. MDM2 inhibits the activity of p53, in part by exporting it to the cytoplasm where it can be degraded by proteasomes, but also by acting as a ubiquitin ligase. p53 becomes activated following proteasome inhibition, which stimulates p53-mediated tumour-suppressor activity; namely, apoptosis and senescence. c | In response to stress, such as neoplasia and chemotherapy, the I B inhibitor becomes phosphorylated and deactivated by the proteasome. Thereafter, nuclear factor of B (NF- B) is expressed and promotes several pro-survival pathways. Proteasome inhibition prevents the activation of NF- B and increases the susceptibility of the cell to cytotoxic effects of chemotherapy. CDK, cyclin-dependent kinase.
 is expressed and promotes several pro-survival pathways. Proteasome inhibition prevents the activation of NF- B and increases the susceptibility of the cell to cytotoxic effects of chemotherapy. CDK, cyclin-dependent kinase.")
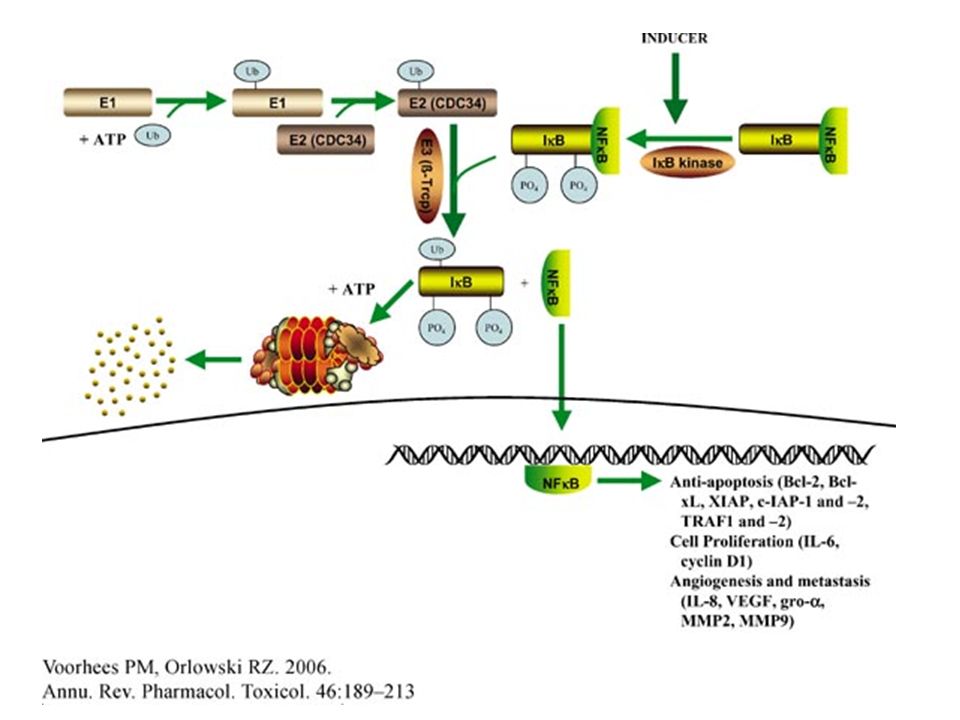
35
Bortezomib and NF- B. A key factor in the ability of the proteasome inhibitor bortezomib to kill myeloma cells is that it blocks the activation of nuclear factor- B (NF- B). In normal cells, NF- B (which exists as a p50–p65 dimer) is bound to the inhibitory protein I B, which maintains it in the inactive form in the cytosol. Certain tumours have activated forms of NF- B, and the proteasome is essential for this activation, as it catalyses the proteolytic generation of the NF- B subunit p50 from the inactive p105 precursor and the destruction of the inhibitory I B. The activated NF- B can then enter the nucleus, which allows it to carry out many functions in the tumour cell that help the cell to survive and proliferate. By inhibiting the proteasome (red cross) and therefore the activation of NF- B (orange crosses), bortezomib helps to reduce anti-apoptotic factors; inflammatory molecules; cell adhesion molecules, which allow attachment cells to adhere to bone marrow cells; and cytokines, which promote the growth of myeloma cells.
. In normal cells, NF- B (which exists as a p50–p65 dimer) is bound to the inhibitory protein I B, which maintains it in the inactive form in the cytosol. Certain tumours have activated forms of NF- B, and the proteasome is essential for this activation, as it catalyses the proteolytic generation of the NF- B subunit p50 from the inactive p105 precursor and the destruction of the inhibitory I B. The activated NF- B can then enter the nucleus, which allows it to carry out many functions in the tumour cell that help the cell to survive and proliferate. By inhibiting the proteasome (red cross) and therefore the activation of NF- B (orange crosses), bortezomib helps to reduce anti-apoptotic factors; inflammatory molecules; cell adhesion molecules, which allow attachment cells to adhere to bone marrow cells; and cytokines, which promote the growth of myeloma cells..")
36
Nuclear factor-κB (NF-κB) is commonly overexpressed in malignant cells, and constitutive activity of NF-κB is frequently observed in different types of cancer. Constitutive activity of NF-κB has been correlated with resistance of cancer cells to radiation- and chemotherapy-induced apoptosis. This constitutive NF-κB activation is caused by the following: alterations affecting genes encoding NF-κB and inhibitor of κB (IκB) that lead to acceleration of NF-κB activation (a); constitutive activation of IκB kinase (IKK) that leads to accelaration of IκB phosphorylation (b); or mutations inactivating IκBs (c).
 that lead to acceleration of NF-κB activation (a); constitutive activation of IκB kinase (IKK) that leads to accelaration of IκB phosphorylation (b); or mutations inactivating IκBs (c)..")
37
Nuclear factor-κB (NF-κB) is commonly overexpressed in malignant cells, and constitutive activity of NF-κB is frequently observed in different types of cancer. Constitutive activity of NF-κB has been correlated with resistance of cancer cells to radiation- and chemotherapy-induced apoptosis. This constitutive NF-κB activation is caused by the following: alterations affecting genes encoding NF-κB and inhibitor of κB (IκB) that lead to acceleration of NF-κB activation (a); constitutive activation of IκB kinase (IKK) that leads to accelaration of IκB phosphorylation (b); or mutations inactivating IκBs (c).
 that lead to acceleration of NF-κB activation (a); constitutive activation of IκB kinase (IKK) that leads to accelaration of IκB phosphorylation (b); or mutations inactivating IκBs (c)..")
38
Nuclear factor-κB (NF-κB) is commonly overexpressed in malignant cells, and constitutive activity of NF-κB is frequently observed in different types of cancer. Constitutive activity of NF-κB has been correlated with resistance of cancer cells to radiation- and chemotherapy-induced apoptosis. This constitutive NF-κB activation is caused by the following: alterations affecting genes encoding NF-κB and inhibitor of κB (IκB) that lead to acceleration of NF-κB activation (a); constitutive activation of IκB kinase (IKK) that leads to accelaration of IκB phosphorylation (b); or mutations inactivating IκBs (c).
 that lead to acceleration of NF-κB activation (a); constitutive activation of IκB kinase (IKK) that leads to accelaration of IκB phosphorylation (b); or mutations inactivating IκBs (c)..")
39
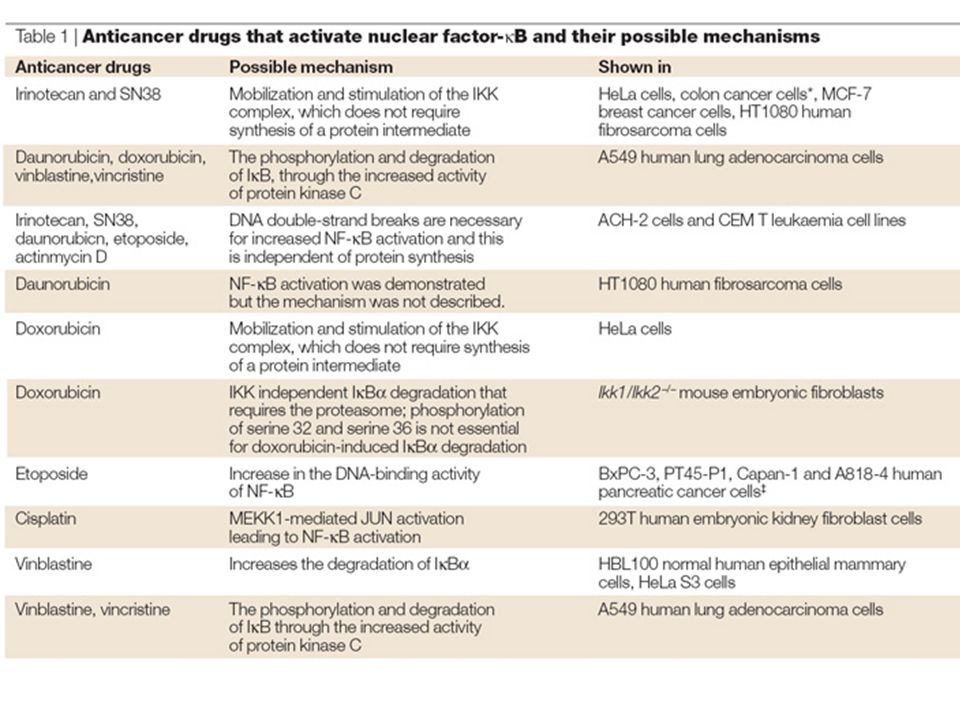
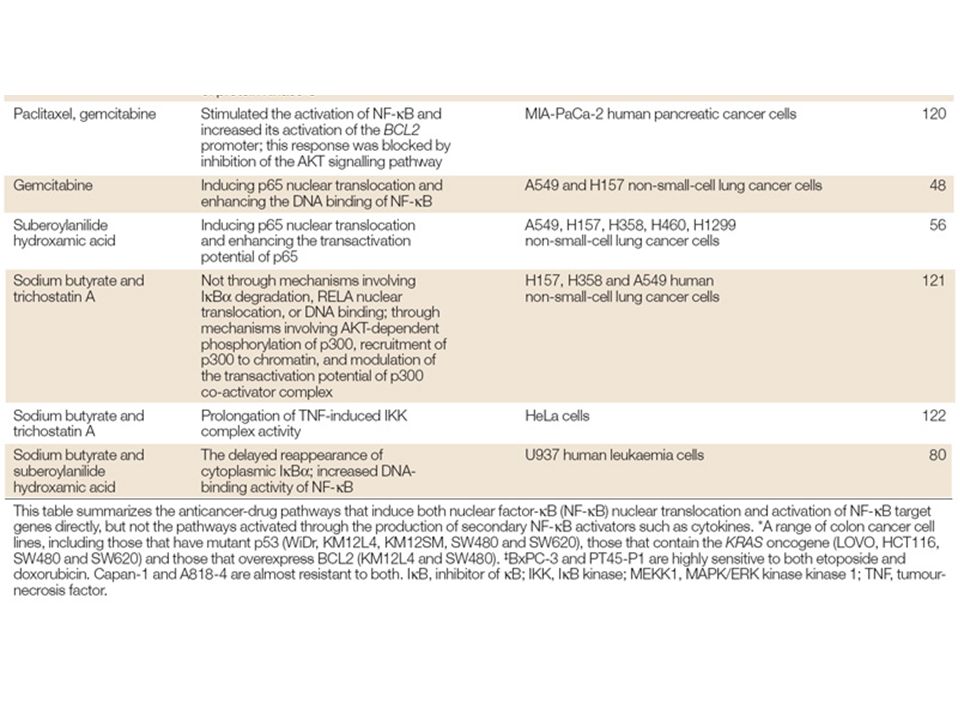
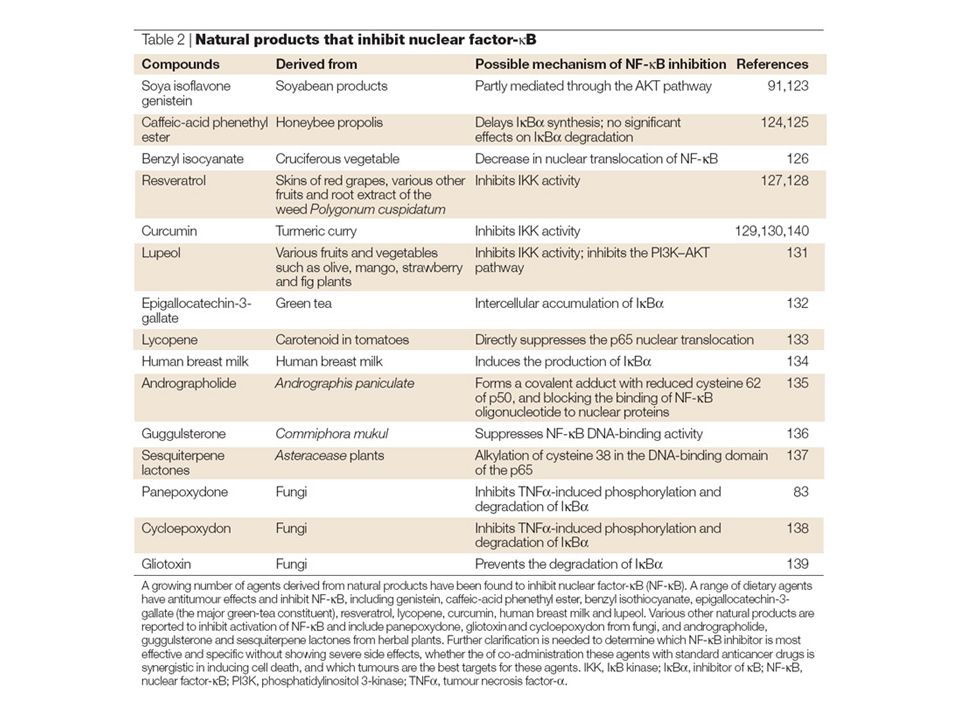
Anticancer drugs induce nuclear translocation of nuclear factor-κ B (NF-κB) and activation of NF-κB target genes through the direct activation of the NF-κB pathway and the secondary production of NF-κB activators such as reactive oxygen species (ROS), interleukin-1 (IL-1) and tumour-necrosis factor-α (TNFα). A number of other factors within the local tumour environment can also activate NF-κB, particularly in the context of an immune response. T-cells activate NF-κB through the activation of the T-cell receptor (TCR) in response to a presented antigen (Ag). All of these effects can lead to enhanced cell proliferation, angiogenesis, motility, migration, adhesion, immortality, inflammation and inhibition of apoptosis, through expression of genes coding for anti-apoptotic proteins, membrane transporters, cytokines, growth factors, cell-cycle regulators, cell-adhesion molecules and cell-surface proteases.The genes encoding cyclooxygenase-2 and inducible nitric oxide synthase 2 (iNOS2) are also induced, which feeds the inflammatory response. 26S, 26S proteasome; BCL2, B-cell CLL/lymphoma 2; COX2, cyclooxygenase-2; GM-CSF, granulocyte-macrophage colony-stimulating factor; IAP, inhibitor of apoptosis protein; ICAM1, intracellular adhesion molecule 1; IEX1L, immediate early response gene X-1 longer transcript; IKK, I B kinase; IL-1Rs, IL-1 receptors; IκB, inhibitor of κB; MDR1, multidrug resistance gene 1; MEKK, MAPK/ERK kinase kinase; MHC, major histocompatibility complex; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB.; PI3K, phosphatidylinositol 3-kinase;TNFR, tumour-necrosis factor receptor; ULS, ubiquitin ligase system; uPA, urokinase-type plasminogen activator; UV, ultraviolet; VCAM1, vascular cell-adhesion molecule 1; VEGF, vascular endothelial growth factor; XIAP, X-linked inhibitor of apoptosis protein.
 in response to a presented antigen (Ag). All of these effects can lead to enhanced cell proliferation, angiogenesis, motility, migration, adhesion, immortality, inflammation and inhibition of apoptosis, through expression of genes coding for anti-apoptotic proteins, membrane transporters, cytokines, growth factors, cell-cycle regulators, cell-adhesion molecules and cell-surface proteases.The genes encoding cyclooxygenase-2 and inducible nitric oxide synthase 2 (iNOS2) are also induced, which feeds the inflammatory response. 26S, 26S proteasome; BCL2, B-cell CLL/lymphoma 2; COX2, cyclooxygenase-2; GM-CSF, granulocyte-macrophage colony-stimulating factor; IAP, inhibitor of apoptosis protein; ICAM1, intracellular adhesion molecule 1; IEX1L, immediate early response gene X-1 longer transcript; IKK, I B kinase; IL-1Rs, IL-1 receptors; IκB, inhibitor of κB; MDR1, multidrug resistance gene 1; MEKK, MAPK/ERK kinase kinase; MHC, major histocompatibility complex; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB.; PI3K, phosphatidylinositol 3-kinase;TNFR, tumour-necrosis factor receptor; ULS, ubiquitin ligase system; uPA, urokinase-type plasminogen activator; UV, ultraviolet; VCAM1, vascular cell-adhesion molecule 1; VEGF, vascular endothelial growth factor; XIAP, X-linked inhibitor of apoptosis protein..")
42
Inhibition of nuclear factor- B (NF- B) results in an increased susceptibility of cancer cells to pro-apoptotic anticancer agents. It seems that the blockade of NF- B activation can shift the tumour survival/death balance towards apoptosis.

44
Sito di sviluppo del tumore
ESEMPI DI INFIAMMAZIONI/INFEZIONI CRONICHE LEGATE A UN AUMENTO DEL RISCHIO DI SVILUPPO DI TUMORI Malattia Sito di sviluppo del tumore Rischio relativo Forme ereditarie Emocromatosi Fegato 219 Malattia di Crohn Colon 3 Colite ulcerosa 6 Forme acquisite Virali Hepatite B 88 Hepatite C 30 Batteriche Helicobacter pylori Stomaco 11 PID (pelvic inflammatory disease) Ovaio Parassitarie S. hematobium Vescica 2-14 S. japonicum 2-6 Fasciola epatica 14 Da cause chimiche/fisiche/metaboliche Riflusso acido Esofago 50-100 Amianto Pleure >10 Obesità Molteplici siti
 Ovaio. Parassitarie. S. hematobium. Vescica S. japonicum Fasciola epatica. 14. Da cause chimiche/fisiche/metaboliche. Riflusso acido. Esofago Amianto. Pleure. >10. Obesità. Molteplici siti")
45
Figure 1. Schematic presentation of a procarcinogenic scenario during chronic inflammation. Generation and accumulation of cytokines, growth factors, free radicals, matrix proteinases and prostaglandins induces several protumorigenic alterations including DNA damage, protein modifications, changes in gene expression profiles and the expression of specific miRNA. Combined with the stromal-epithelial interaction and immune suppressive effect of adaptive immune response, these changes can support tumor development and invasion. International Journal of Cancer Volume 121, Issue 11, Pages

46
Figure 2. Chronic inflammation affects several crucial pathways involved in the maintenance of cellular homeostasis. Free radicals generated during inflammation cause DNA damage leading to the activation of a p53 stress response pathway and the inactivation of the pRb tumor suppressive pathway through their post-translational modifications. However, the oncogenic pathway, e.g., the activation of NF- B, also occurs along with the inactivation or oncogenic transformation of p53 gene due to missense mutations affecting DNA repair, cell cycle arrest and apoptosis.

47
Figure 3. Several reactive oxygen (ROS) and reactive nitrogen species (RNS) are generated during chronic inflammation. The reactive species can induce DNA damage, including point mutations in cancer-related genes, and modifications in essential cellular proteins that are involved in DNA repair, apoptosis and cell cycle, either directly or indirectly through the activation of lipid peroxidation and generation of reactive aldehydes, e.g., malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE).
 and reactive nitrogen species (RNS) are generated during chronic inflammation. The reactive species can induce DNA damage, including point mutations in cancer-related genes, and modifications in essential cellular proteins that are involved in DNA repair, apoptosis and cell cycle, either directly or indirectly through the activation of lipid peroxidation and generation of reactive aldehydes, e.g., malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE)..")
48
Figure 4. NO possesses both protumorigenic and antitumorigenic properties. It can activate the protective p53 stress response pathway and cause oncogenic p53 mutations

49
Figure 5. Examples of cytokine-miRNA interactions in inflammation.

50
Figure 6. Both NOS2 and COX2 cooperate in colon carcinogenesis
Figure 6. Both NOS2 and COX2 cooperate in colon carcinogenesis. The inflammation and Wnt-signaling can induce NOS2 and COX2 leading to the generation of a high level of NO and PGE2, respectively. NO and reactive aldehydes cause DNA alterations including p53 mutations causing a selective growth advantage of the p53 mutant cells. PGE2, in addition to inhibiting apoptosis, can further enhance COX2 production through the activation of the Wnt-pathway. However, NO -induced post-translational modification of p53 can activate p53, which can inhibit NOS2 and inhibit or activate COX2. Furthermore, NO can induce and activate COX2. The ultimate effect of these interactions can lead to genomic instability and cancer.

Presentazioni simili

















 SPECIFICA/SELETTIVA:>")


>")
