Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Meccanismi di tossicità
Meccanismi Meccanismi specifici non specifici Tossine animali Composti organici Composti e vegetali di sintesi inorganici Farmaci

2
Meccanismi specifici Interazione selettiva con specifiche proteine (enzimi, canali ionici, trasportatori ecc.) danneggiamento di funzioni cellulari specifiche tossicità cellulare e/o d’organo. In genere legame non covalente. Razionale biologico?: selezionate sostanze non tossiche per l’organismo che le produce; a scopo difensivo, l’effetto tossico deve essere intenso e manifestarsi a breve termine.
 danneggiamento di funzioni cellulari specifiche tossicità cellulare e/o d’organo. In genere legame non covalente. Razionale biologico : selezionate sostanze non tossiche per l’organismo che le produce; a scopo difensivo, l’effetto tossico deve essere intenso e manifestarsi a breve termine.")
3
Meccanismi non specifici
La sostanza o un suo metabolita reattivo (es.: radicale libero, elettrofilo), reagiscono in modo aspecifico con diversi componenti cellulari (proteine, lipidi, acidi nucleici). Si formano o si rompono legami covalenti (formazione di addotti; ossidazioni; sottrazione di atomi). Ciò causa alterato funzionamento o distruzione dei componenti cellulari colpiti tossicità cellulare tossicità di organo/sistema. In genere, non è chiaro quali dei numerosi siti di attacco cellulare siano responsabili dell’effetto tossico.
, reagiscono in modo aspecifico con diversi componenti cellulari (proteine, lipidi, acidi nucleici). Si formano o si rompono legami covalenti (formazione di addotti; ossidazioni; sottrazione di atomi). Ciò causa alterato funzionamento o distruzione dei componenti cellulari colpiti tossicità cellulare tossicità di organo/sistema. In genere, non è chiaro quali dei numerosi siti di attacco cellulare siano responsabili dell’effetto tossico.")
4
Questo tipo di meccanismo è dovuti quasi sempre alla formazione di metaboliti reattivi e non alla sostanza tal quale. Perché?

5
Alcune sostanze modificano la composizione dei fluidi biologici:
pH (acidi e basi); composizione ionica (sali, chelanti); cofattori enzimatici (deplezione, malassorbimento) concentrazione di metaboliti intermedi (stimolazione o inibizione di vie metaboliche; es.: porfiria, steatosi) Queste alterazioni causano tossicità cellulare e/o d’organo.
; composizione ionica (sali, chelanti); cofattori enzimatici (deplezione, malassorbimento) concentrazione di metaboliti intermedi (stimolazione o inibizione di vie metaboliche; es.: porfiria, steatosi) Queste alterazioni causano tossicità cellulare e/o d’organo.")
6
N.B.: oltre a tossine che agiscono in modo selettivo, i vegetali contengono anche molti composti che danno tossicità con meccanismo non specifico. Molti farmaci e alcuni altri composti organici di sintesi danno tossicità con meccanismo specifico.

7
Tipi di tossicità Tossicità funzionale: alterazioni delle funzioni di un sistema (nervoso, cardiovascolare, endocrino) possibile danno d’organo. Citotossicità: alterazione irreversibile di proteine (enzimi, canali ionici ecc.) o membrane (lipidi); disregolazione del metabolismo necrosi, apoptosi tossicità tissutale e d’organo o sistema (fegato, rene, SNC ecc.). Genotossicità: alterazione del DNA mutazioni, alterazioni cromosomiche cancerogenesi, malattie congenite. Reazioni allergiche: allergeni, apteni, modificatori degli antigeni cellulari risposta immunitaria.
 o membrane (lipidi); disregolazione del metabolismo necrosi, apoptosi tossicità tissutale e d’organo o sistema (fegato, rene, SNC ecc.). Genotossicità: alterazione del DNA mutazioni, alterazioni cromosomiche cancerogenesi, malattie congenite. Reazioni allergiche: allergeni, apteni, modificatori degli antigeni cellulari risposta immunitaria.")
8
1. Alterazione delle normali funzioni cellulari senza danno cellulare primario diretto (tossicità ‘funzionale’) tossicità d’organo o sistema (tossicità cellulare secondaria) Esempi: Una sostanza che provochi vasodilatazione (es., alfa1-bloccante, agonista istaminergico) non causa danni diretti ai vasi ma provoca ipotensione e riduzione del flusso ematico danni cerebrali, renali ecc Una sostanza che causa contrazione della muscolatura vasale (es. agonista adrenergico) non danneggia i vasi ma causa tossicità cardiaca, cerebrale, renale.
 non causa danni diretti ai vasi ma provoca ipotensione e riduzione del flusso ematico danni cerebrali, renali ecc. Una sostanza che causa contrazione della muscolatura vasale (es. agonista adrenergico) non danneggia i vasi ma causa. tossicità cardiaca, cerebrale, renale.")
9
Una sostanza che stimoli il rilascio di insulina dalle cellule pancreatiche (senza danneggiarle) causa ipoglicemia e tossicità a carico del sistema nervoso centrale (danno neuronale indiretto) Una sostanza antagonista degli ormoni sessuali può causare tossicità riproduttiva senza causare danno diretto alle cellule dell’apparato riproduttivo
 Una sostanza antagonista degli ormoni sessuali può causare. tossicità riproduttiva senza causare danno diretto alle cellule dell’apparato riproduttivo.")
10
Danno cellulare (citotossicità).
L’alterato funzionamento o la distruzione della macromolecola bersaglio provoca una danno cellulare L’entità di tale danno dipende da: importanza della funzione del componente cellulare colpito capacità di riparazione da parte della cellula (rimozione dei componenti danneggiati e loro sostituzione )
")
11
citotossicità Se il danno è irreversibile e/o esteso e/o coinvolge componenti cellulari essenziali, si ha la morte cellulare (necrosi). Nella maggior parte dei tessuti, le cellule morte possono essere sostituite da cellule dello stesso tipo (divisione cellulare) riparazione totale del danno. N.B.: i neuroni non possono replicarsi. Se l’area necrotica è estesa, si ha infiammazione e formazione di tessuti connettivi di riparazione (tessuti cicatriziali) riparazione parziale del danno (la funzionalità dell’organo è diminuita o alterata).
 riparazione totale del danno. N.B.: i neuroni non possono replicarsi. Se l’area necrotica è estesa, si ha infiammazione e formazione di tessuti connettivi di riparazione (tessuti cicatriziali) riparazione parziale del danno (la funzionalità dell’organo è diminuita o alterata).")
12
In questi casi si possono avere alterazioni di:
citotossicità Se l’insulto cellulare non è ‘grave’, si può avere alterazione del metabolismo della cellula senza morte cellulare. In questi casi si possono avere alterazioni di: dimensioni cellulari (atrofia o ipertrofia) proliferazione e differenziazione cellulare (iperplasia, metaplasia) accumulo di componenti cellulari (es. steatosi epatica)
 proliferazione e differenziazione cellulare (iperplasia, metaplasia) accumulo di componenti cellulari (es. steatosi epatica)")
13
La morte cellulare può avvenire per necrosi o per apoptosi
citotossicità La morte cellulare può avvenire per necrosi o per apoptosi

14
Necrosis Apoptosis Stimuli Histology DNA Breakdown Tissue Reaction
Necrosis Apoptosis Stimuli Pathologic (hypoxia, toxins,etc.). Consequence of irreversible cell injury. Think of necrosis as "cell homicide". A physiologic, genetically regulated process. Occasionally activated by pathologic stimuli. Think of apoptosis as "cell suicide". Histology - Typically large numbers of cells affected - Cell swelling. - Cellular acidosis. - Organelle disruption. - Loss of membrane integrity. - Coagulation or liquefaction of cell proteins. - Usually only a few cells affected. - Cell shrinkage due to hydrolysis and cross-linking of structural proteins within the cytoplasm and nucleus. - Organelles remain normal. - Cell breaks down into membrane-bound fragments (apoptotic bodies) which are taken up by neighboring cells. DNA Breakdown Random, diffuse fragmentation and dissolution of the nucleus. Orderly nuclear condensation and fragmentation. Tissue Reaction Inflammation with secondary injury to surrounding normal tissues. No Inflammation or secondary tissue injury.
. Consequence of irreversible cell injury. Think of necrosis as cell homicide . A physiologic, genetically regulated process. Occasionally activated by pathologic stimuli. Think of apoptosis as cell suicide . Histology. - Typically large numbers of cells affected - Cell swelling. - Cellular acidosis. - Organelle disruption. - Loss of membrane integrity. - Coagulation or liquefaction of cell proteins. - Usually only a few cells affected. - Cell shrinkage due to hydrolysis and cross-linking of structural proteins within the cytoplasm and nucleus. - Organelles remain normal. - Cell breaks down into membrane-bound fragments (apoptotic bodies) which are taken up by neighboring cells. DNA Breakdown. Random, diffuse fragmentation and dissolution of the nucleus. Orderly nuclear condensation and fragmentation. Tissue Reaction. Inflammation with secondary injury to surrounding normal tissues. No Inflammation or secondary tissue injury.")
15
citotossicità

16
Siti di attacco cellulare
citotossicità Siti di attacco cellulare Membrane: Lipidi: perossidazione Proteine: enzimi, canali, pompe, trasportatori: alterazione della funzionalità

17
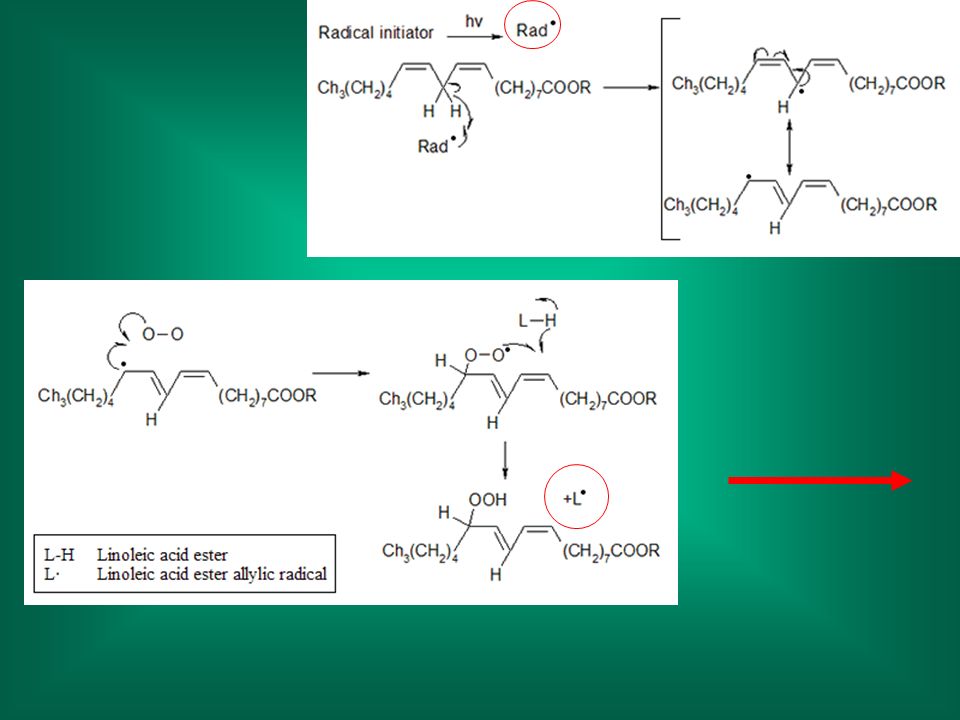
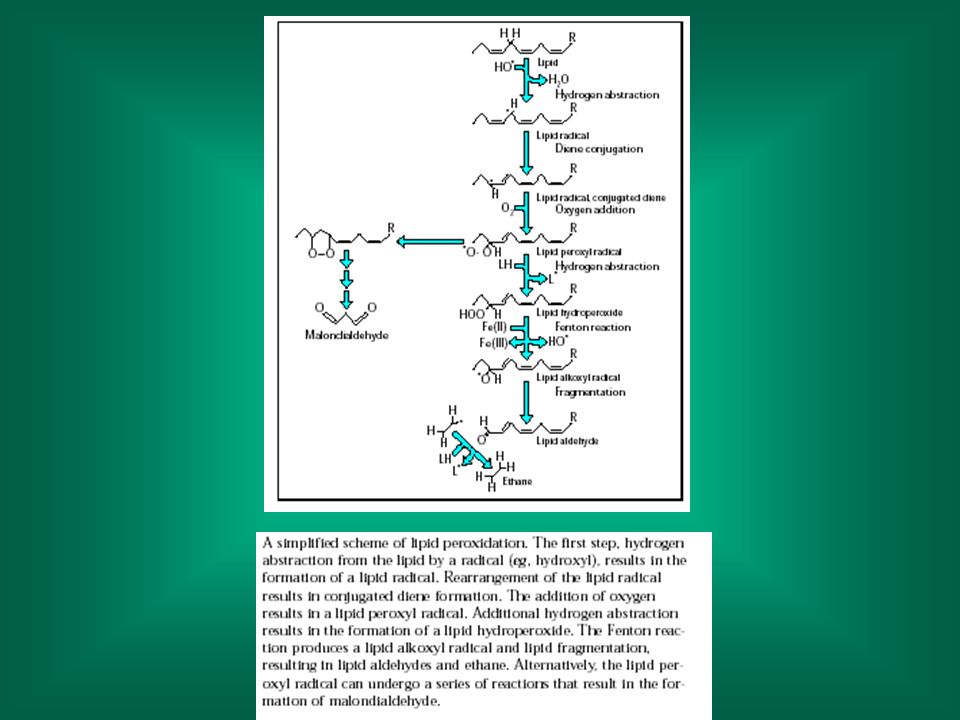
Perossidazione lipidica
citotossicità: danno alle membrane Perossidazione lipidica Avviene ad opera di radicali (liberi): meccanismo non specifico Dato che i radicali hanno molti altri punti bersagli cellulari (proteine, DNA), il suo ruolo nel danno cellulare in molti casi non è ben definito (causa o effetto?)
: meccanismo non specifico. Dato che i radicali hanno molti altri punti bersagli cellulari (proteine, DNA), il suo ruolo nel danno cellulare in molti casi non è ben definito (causa o effetto )")
20
La perossidazione porta alla distruzione dei lipidi alterazione della struttura della membrana alterazioni della funzionalità. I lipidi ossidati possono a loro volta ossidare le proteine di membrana. La formazione di radicali può danneggiare strutture distanti dal sito iniziale d’attacco.

21
Nella perossidazione lipidica si formano prodotti di degradazione;
questi sono utilizzati per misurare il grado di perossidazione; in genere si misura la formazione della malonildialdeide (dialdeide malonica)
")
22
CCl4

23
Esempio; valutazione dell’effetto ossidativo in ratti trattati con CCl4 (s.c.) per 7 settimane
Oxidative stress markers Mean renal MDA (malondialdehyde) and GSH-Px (glutathione peroxidase) levels in the control, CCl4 and CCl4+INF groups GSH-Px (u/mg protein) MDA (nmol/mg protein) Control 45.4±5.9 4.15±0.5 CCl4 34.5±3.1* 5.55±0.7* CCl4+Interferon 46.5±6.4** 4.27±0.3** *p<0.01 vs. control, **p>0.05 vs. control.
 and GSH-Px (glutathione peroxidase) levels in the control, CCl4 and CCl4+INF groups. GSH-Px (u/mg protein) MDA (nmol/mg protein) Control. 45.4± ±0.5. CCl ±3.1* 5.55±0.7* CCl4+Interferon. 46.5±6.4** 4.27±0.3** *p<0.01 vs. control, **p>0.05 vs. control.")
24
Sbilanciamento dei sistemi redox cellulari
citotossicità Sbilanciamento dei sistemi redox cellulari La cellula ha numerosi sistemi redox, necessari al corretto metabolismo (GSH/GSSG, NAD/NADH, NADP/NADPH, piruvato/lattato ecc.). L’attività di molte proteine (enzimi, fattori di trascrizione ecc.) è regolata dallo stato redox della cellula.
. L’attività di molte proteine (enzimi, fattori di trascrizione ecc.) è regolata dallo stato redox della cellula.")
25
Regolazione dell’attività di una monoossigenasi flavinica da parte dello stato redox (GSH/GSSG)
")
26
Figure 1. Diagram of the NF-kB activation pathway indicating the steps where there is evidence for direct modulation by reactive oxygen species (ROS)
.")
27
La diminuzione di GSH comporta, tra l’altro, una diminuita protezione nei confronti dell’ossidazione dei tioli proteici alterazioni strutturali e funzionali. La detossificazione di ossidanti e radicali comporta un consumo di equivalenti riducenti ed un conseguente sbilanciamento dei sistemi redox alterazioni del metabolismo cellulare.

28
citotossicità L’alterazione dei sistemi redox può anche causare effetti tossici indirettamente. Es.: la detossificazione di H2O2 da parte della glutatione perossidasi porta alla formazione di glutatione ossidato (GSSG); la rigenerazione di glutatione ridotto ad opera della glutatione reduttasi comporta il consumo di NADPH alterazione del rapporto NAPD/NADPH alterazione di molte funzioni cellulari (es. efflusso di Ca++ dai mitocondri).
; la rigenerazione di glutatione ridotto ad opera della glutatione reduttasi comporta il consumo di NADPH alterazione del rapporto NAPD/NADPH alterazione di molte funzioni cellulari (es. efflusso di Ca++ dai mitocondri).")
30
Anche lo sbilanciamento verso lo stato ridotto altera il metabolismo.
citotossicità Anche lo sbilanciamento verso lo stato ridotto altera il metabolismo. Ad esempio, l’etanolo viene ossidato in due stadi ad acetato: etanolo + NAD acetaldeide + NADH acetaldeide + NAD acetato + NADH Negli alcolisti si ha quindi una sovrapproduzione di NADH, che favorisce la riduzione di piruvato a lattato e di ossalacetato a malato, diminuendo così la gluconeogenesi. Il risultato finale è una condizione di ipoglicemia.

32
Disregolazione metabolica. Principali meccanismi
citotossicità Disregolazione metabolica. Principali meccanismi Inibizione del metabolismo energetico (produzione di ATP) Aumento del Ca++ libero intracellulare Alterazione della sintesi di macromolecole (acidi nucleici, proteine)
 Aumento del Ca++ libero intracellulare. Alterazione della sintesi di macromolecole (acidi nucleici, proteine)")
33
Inibizione della produzione di ATP
citotossicità Inibizione della produzione di ATP Può avvenire per blocco della glicolisi (es. iodoacetato) o della respirazione mitocondriale (es., cianuro, disaccoppianti)
 o della respirazione mitocondriale (es., cianuro, disaccoppianti)")
34
Il gradiente di H+ viene utilizzato per generare ATP.
I disaccoppianti provocano un trasporto di H+ passivo attraverso la membrana inibizione della sintesi di ATP.

35
CN- si lega al Fe+3 del gruppo eme blocco del flusso degli elettroni

36
Inibizione totale necrosi cellulare
citotossicità Inibizione totale necrosi cellulare Inibizione parziale e/o temporanea riduzione del metabolismo, ridotta capacità rigenerativa, aumentata sensibilità a stimoli tossici

37
Aumento del Ca++ intracellulare
citotossicità Aumento del Ca++ intracellulare La concentrazione di Ca++ intracellulare libero è strettamente regolata poiché Ca++ è necessario per l’attività di molti enzimi (proteasi, fosfolipasi, endonucleasi ecc.) e regola molte attività cellulari Sostanze che danneggiano i sistemi responsabili del mantenimento della concentrazione di Ca++ provocano un suo aumento con attivazione di molti enzimi litici e conseguente morte cellulare (necrosi o apoptosi).
 e regola molte attività cellulari. Sostanze che danneggiano i sistemi responsabili del mantenimento della concentrazione di Ca++ provocano un suo aumento con attivazione di molti enzimi litici e conseguente morte cellulare (necrosi o apoptosi).")
38
proteasi Ca-dipendenti
proteasi lisosomiali

39
Sinergismo in tossicologia
Molto importante per esposizione contemporanea a molte sostanze diverse. L’esposizione ad una sostanza, in modalità non tossiche, determina un aumento della tossicità di una seconda sostanza

40
Acetaminophen Overdose
Dose related toxicity in low risk patient: < 125 mg/kg (8.75 g in 70 kg person) - rare liver toxicity >250 mg/kg (17.5 g) considered "minimum hepatotoxic dose" > 350 mg/kg (24.5 g) -- invariably severe liver damage High risk patient: In chronic ethanol abusers, hepatic necrosis has been reported with short-term use of paracetamol dosages of 2.5 g/day. Fatalities have occurred.
 - rare liver toxicity. >250 mg/kg (17.5 g) considered minimum hepatotoxic dose > 350 mg/kg (24.5 g) -- invariably severe liver damage. High risk patient: In chronic ethanol abusers, hepatic necrosis has been reported with short-term use of paracetamol dosages of 2.5 g/day. Fatalities have occurred.")
42
Reazioni allergiche e autoimmuni indotte da metaboliti reattivi
Diverse sostanze possono modulare in modo aspecifico il sistema immunitario, determinando immunosoppressione (più comune) o immunostimolazione generalizzata (più raro, es.; silicosi, esaclorobenzene) Molte sostanze si comportano da allergeni o apteni, inducendo una risposta immunitaria reazioni allergiche (Tipo I-IV). I metaboliti reattivi di alcune sostanze si legano covalentemente ad alcune proteine, modificandone le caratteristiche immunitarie malattia autoimmunitaria (es. epatite da alotano).
 o immunostimolazione generalizzata (più raro, es.; silicosi, esaclorobenzene) Molte sostanze si comportano da allergeni o apteni, inducendo una risposta immunitaria reazioni allergiche (Tipo I-IV). I metaboliti reattivi di alcune sostanze si legano covalentemente ad alcune proteine, modificandone le caratteristiche immunitarie malattia autoimmunitaria (es. epatite da alotano).")
43
Formazione di composti tossici in vivo. Meccanismi di detossificazione
Formazione di radicali. Per ossidazione di fenoli, idrochinoni, amine, idrazine. tioli ecc. da parte di CYP450 o perossidasi: R R.+ + 1e si forma una radicale cationico, che può sottrarre un atomo di idrogeno a lipidi ed altre molecole (proteine, DNA, glutatione ecc.). Per scissione omolitica di un legame C-H; es: CCl4 subisce dealogenazione riduttiva ad opera di CYP450; si forma il radicale triclorometilico CCl3.
. Per scissione omolitica di un legame C-H; es: CCl4 subisce dealogenazione riduttiva ad opera di CYP450; si forma il radicale triclorometilico CCl3.")
44
Per riduzione ad opera della CYP450 reduttasi (e altre reduttasi): R + 1e R.- (radicale anionico); es.: doxorubicina, nitrofurantoina. I radicali anionici cedono il loro elettrone spaiato all’O2, formando cosi l’anione superossido: R.- + O2 R + O2 .-

45
L’anione superossido viene trasformato dalla superossido dismutasi (SOD) in perossido di idrogeno (H2O2); H2O2 (un ossidante) viene detossificato dalla catalasi: H2O2 H2O + ½ O2 Tuttavia, la formazione di anione superossido e/o di perossido di idrogeno può superare le capacità detossificanti di SOD e catalasi. H2O2 può ricevere un elettrone da ioni metallici (Fe++, Cr(V) ecc.), formando il radicale ossidrile (reazione di Fenton): H2O2 OH. + OH- Il radicale OH. è estremamente reattivo e non può essere inattivato dagli antiossidanti
 viene detossificato dalla catalasi: H2O2 H2O + ½ O2. Tuttavia, la formazione di anione superossido e/o di perossido di idrogeno può superare le capacità detossificanti di SOD e catalasi. H2O2 può ricevere un elettrone da ioni metallici (Fe++, Cr(V) ecc.), formando il radicale ossidrile (reazione di Fenton): H2O2 OH. + OH- Il radicale OH. è estremamente reattivo e non può essere inattivato dagli antiossidanti.")
46
Anione superossido, perossido di idrogeno e radicale ossidrile vengono definiti ROS (Reactive Oxygen Species)
")
47
I ROS sono prodotti continuamente nel nostro organismo
I ROS sono prodotti continuamente nel nostro organismo. Ad esempio la reazione: Hypoxanthine + O2 <=> Xanthine + H2O2 (catalizzata dalla xantina ossidasi) genera perossido di idrogeno I ROS prodotti dalle cellule fagocitarie svolgono un ruolo nella difesa dagli agenti infettivi. I radicali possono danneggiare anche proteine (principalmente ossidazione dei tioli) e DNA
 genera perossido di idrogeno. I ROS prodotti dalle cellule fagocitarie svolgono un ruolo nella difesa dagli agenti infettivi. I radicali possono danneggiare anche proteine (principalmente ossidazione dei tioli) e DNA.")
48
Meccanismi di detossificazione dei ROS
Protezione enzimatica Glutatione perossidasi H2O2 + 2 GSH H2O + GSSG Glutatione ridotto Glutatione ossidato

49
Altri meccanismi di detossificazione dei radicali
Il glutatione ridotto (GSH) reagisce con i radicali, cedendo un elettrone; si forma così il radicale tiilico GS. (relativamente stabile); l’unione di 2 radicali GS. porta alla formazione di glutatione ossidato (GSSG), che viene poi ridotto dalla glutatione reduttasi (NADPH-dipendente)
 reagisce con i radicali, cedendo un elettrone; si forma così il radicale tiilico GS. (relativamente stabile); l’unione di 2 radicali GS. porta alla formazione di glutatione ossidato (GSSG), che viene poi ridotto dalla glutatione reduttasi (NADPH-dipendente)")
50
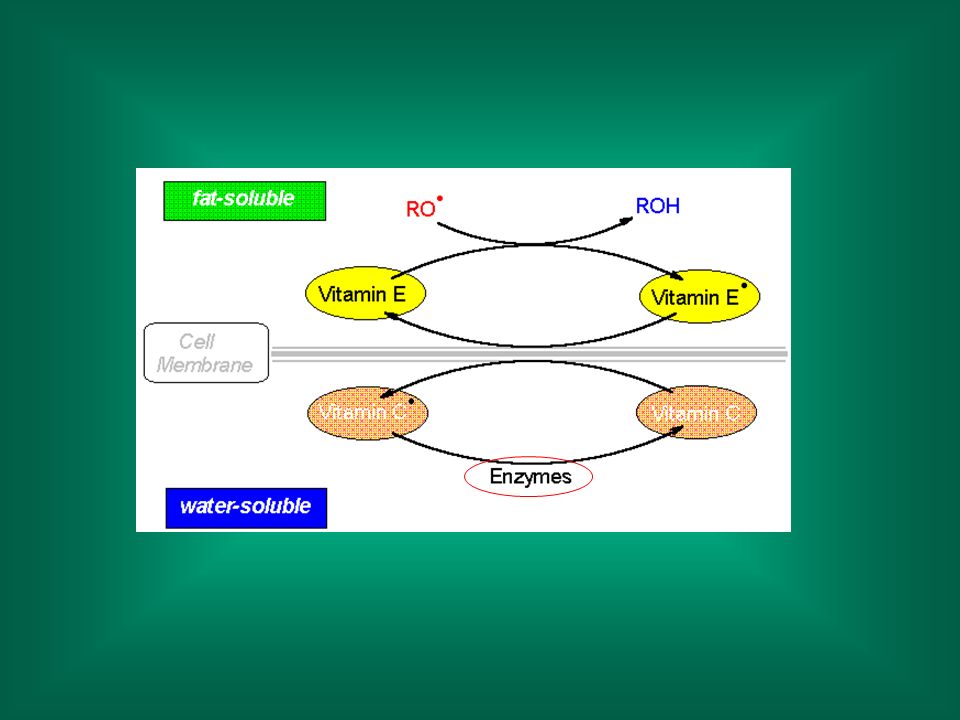
Antiossidanti Vitamina E (alfa-tocoferolo), vitamina C (acido ascorbico) ed altri antiossidanti (es.: ß-carotene) reagiscono con i radicali liberi, cedendo un elettrone e trasformandosi in radicali ‘stabili’ o detossificati dai sistemi enzimatici.
, vitamina C (acido ascorbico) ed altri antiossidanti (es.: ß-carotene) reagiscono con i radicali liberi, cedendo un elettrone e trasformandosi in radicali ‘stabili’ o detossificati dai sistemi enzimatici.")
51
La reazione del radicale perossilico con la Vitamina E è molto più veloce della reazione con un altro lipide (109 vs 106 nmoli/sec)
")
52
Membrane lipid peroxidation
Membrane lipid peroxidation. (a) Initiation of the peroxidation process by an oxidizing radical X · , by abstraction of a hydrogen atom, thereby forming a pentadienyl radical. (b) Oxygenation to form a peroxyl radical and a conjugated diene. (c) Peroxyl radical moiety partitions to the water-membrane interface where it is poised for repair by tocopherol. (d) Peroxyl radical is converted to a lipid hydroperoxide, and the resulting tocopherol radical can be repaired by ascorbate. (e) Tocopherol has been recycled by ascorbate; the resulting ascorbate radical can be recycled by enzyme systems. The enzymes phospholipase A2 (PLA2), phospholipid hydroperoxide glutathione peroxidase (PH-GPx), glutathione peroxidase (GPx) and fatty acyl-coenzyme A (FA-CoA) cooperate to detoxify and repair the oxidized fatty acid chain of the phospholipid. (from Buettner 1993).
 Initiation of the peroxidation process by an oxidizing radical X · , by abstraction of a hydrogen atom, thereby forming a pentadienyl radical. (b) Oxygenation to form a peroxyl radical and a conjugated diene. (c) Peroxyl radical moiety partitions to the water-membrane interface where it is poised for repair by tocopherol. (d) Peroxyl radical is converted to a lipid hydroperoxide, and the resulting tocopherol radical can be repaired by ascorbate. (e) Tocopherol has been recycled by ascorbate; the resulting ascorbate radical can be recycled by enzyme systems. The enzymes phospholipase A2 (PLA2), phospholipid hydroperoxide glutathione peroxidase (PH-GPx), glutathione peroxidase (GPx) and fatty acyl-coenzyme A (FA-CoA) cooperate to detoxify and repair the oxidized fatty acid chain of the phospholipid. (from Buettner 1993).")
54
E’ possibile aumentare la capacità detossificante tramite la somministrazione di antiossidanti?
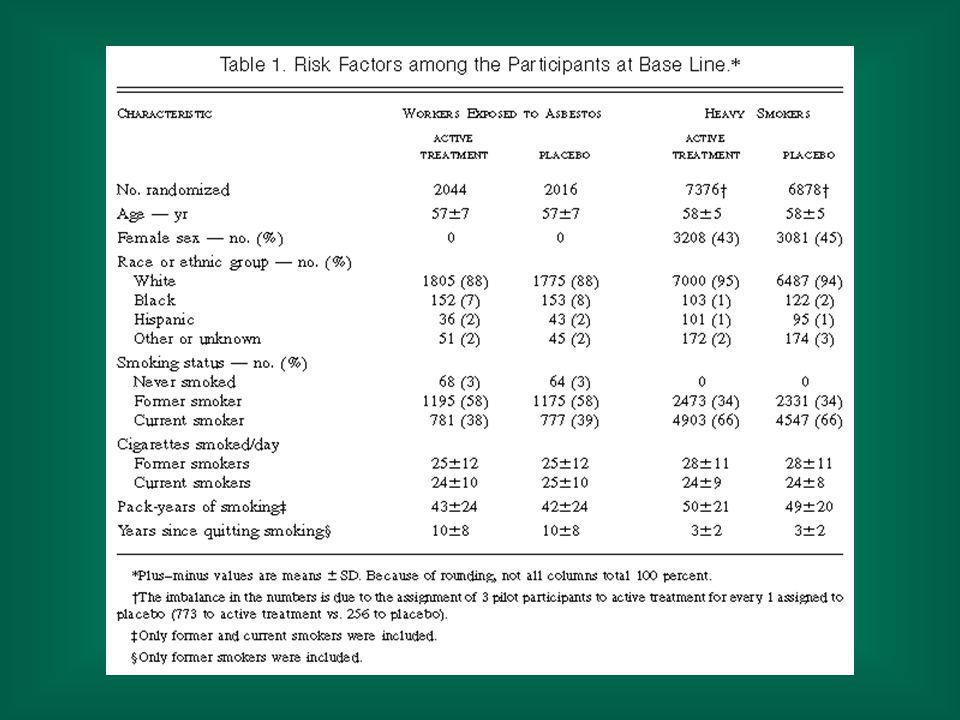
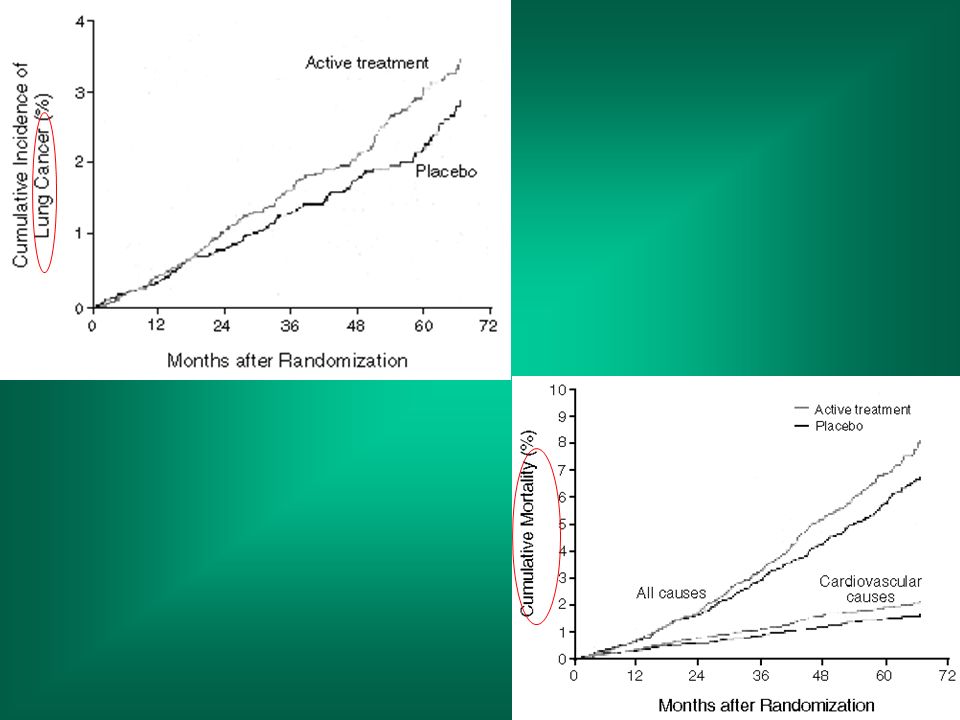
NEJM, Vol. 334: May 2, 1996 Number 18 Effects of a Combination of Beta Carotene and Vitamin A on Lung Cancer and Cardiovascular Disease Gilbert S. Omenn, M.D., Ph.D., Gary E. Goodman, M.D., M.S., Mark D. Thornquist, Ph.D., John Balmes, M.D., Mark R. Cullen, M.D., Andrew Glass, M.D., James P. Keogh, M.D., Frank L. Meyskens, M.D., Barbara Valanis, Dr.P.H., James H. Williams, M.D., Scott Barnhart, M.D., M.P.H., and Samuel Hammar, M.D.

55
Methods We conducted a multicenter, randomized, double-blind, placebo-controlled primary prevention trial — the Beta-Carotene and Retinol Efficacy Trial — involving a total of 18,314 smokers, former smokers, and workers exposed to asbestos. The effects of a combination of 30 mg of beta carotene per day and 25,000 IU of retinol (vitamin A) in the form of retinyl palmitate per day on the primary end point, the incidence of lung cancer, were compared with those of placebo.
 in the form of retinyl palmitate per day on the primary end point, the incidence of lung cancer, were compared with those of placebo.")
58
Formazione di elettrofili
Si formano direttamente per ossidazione, principalmente da parte di CYP450 (es. epossidi di alcheni o aromatici), o per ossidazione e successivo riarrangiamento (es. N-ossidazione di amine).
, o per ossidazione e successivo riarrangiamento (es. N-ossidazione di amine).")
59
Attivazione metabolica: formazione di metaboliti reattivi da parte del CYP450
L’ossidazione da parte del CYP450 può portare alla formazione di composti elettrofili o radicalici (o loro precursori). Epossidi Chinoni, idrochinoni, chinonimmine N-idrossi ammine Radicali alchilici alogenati N.B. non tutti i composti di queste classi sono tossici; la loro tossicità dipende dalla loro reattività chimica, che dipende dalla struttura dell’intera molecola. N.B. in diversi casi (es. aflatossina B1), il CYP450 catalizza sia l’attivazione sia la detossificazione
. Epossidi. Chinoni, idrochinoni, chinonimmine. N-idrossi ammine. Radicali alchilici alogenati. N.B. non tutti i composti di queste classi sono tossici; la loro tossicità dipende dalla loro reattività chimica, che dipende dalla struttura dell’intera molecola. N.B. in diversi casi (es. aflatossina B1), il CYP450 catalizza sia l’attivazione sia la detossificazione.")
60
Gli elettrofili ed i radicali cationici si legano covalentemente ai numerosi gruppi nucleofili presenti in macromolecole, proteine ed acidi nucleici. La reazione con le proteine può portare ad alterazioni della loro funzionalità (es. inibizione enzimatica) e delle loro caratteristiche antigeniche (patologie autoimmuni da xenobiotici). La reazione con il DNA può portare a mutazioni cancerogenesi. La reattività degli elettrofili verso i nucleofili è determinata dalla loro natura: elettrofili soft reagiscono preferenzialmente con nucleofi soft, elettrofili hard con nucleofili hard
 e delle loro caratteristiche antigeniche (patologie autoimmuni da xenobiotici). La reazione con il DNA può portare a mutazioni cancerogenesi. La reattività degli elettrofili verso i nucleofili è determinata dalla loro natura: elettrofili soft reagiscono preferenzialmente con nucleofi soft, elettrofili hard con nucleofili hard.")
61
Se l’ammina viene N-idrossilata (da CYP450) prima di essere acetilata, la NAT può, in questo caso, catalizzare una O-acetilazione. Il gruppo acetossi che si forma è un buon ‘gruppo uscente’. Si può formare uno ione nitrenio, elettrofilo altamente reattivo.

62
Meccanismi di detossificazione degli elettrofili
Coniugazione con glutatione. Il gruppo tiolico del glutatione è nucleofilo. La reazione può essere spontanea o catalizzata dalla glutatione-S-transferasi (enzima di fase II). Detossificazione enzimatica: epossido idrolasi; riduzione enzimatica (es. chinoni, DT-diaforasi); ossidazione enzimatica (es. aldeidi, aldeide deidrogenasi)
. Detossificazione enzimatica: epossido idrolasi; riduzione enzimatica (es. chinoni, DT-diaforasi); ossidazione enzimatica (es. aldeidi, aldeide deidrogenasi)")
63
‘Fallimento’ dei meccanismi di detossificazione
Saturazione del potere detossificante: saturazione enzimatica, consumo dei coenzimi, deplezione delle molecole protettive (glutatione, es. paracetamolo) Reversibilità delle reazioni di coniugazione (es. naftilammine; i glucuronidi sono idrolizzati nel rene rilascio del composto ossidato formazione di metaboliti elettrofili tossici) In alcune reazioni di detossificazione si possono formare composti tossici (es. GS.)
 Reversibilità delle reazioni di coniugazione (es. naftilammine; i glucuronidi sono idrolizzati nel rene rilascio del composto ossidato formazione di metaboliti elettrofili tossici) In alcune reazioni di detossificazione si possono formare composti tossici (es. GS.)")
64
Meccanismi di riparazione
Riparazione molecolare: proteine: degradazione e neosintesi; riparazione enzimatica di specifici gruppi (es. riduzione di ponti disolfuro) lipidi: idrolisi + riduzione dell’acido grasso perossidato (ossidazione del glutatione); ri-acilazione
 lipidi: idrolisi + riduzione dell’acido grasso perossidato (ossidazione del glutatione); ri-acilazione.")
65
Riparazione del DNA Il DNA subisce continuamente danni di vario tipo
Esistono diversi meccanismi di riparazione, che sono essenziali al mantenimento dell’integrità del DNA I meccanismi di riparazione richiedono tempo Il danno al DNA determina, tra l’altro, un arresto della progressione del ciclo cellulare, in modo da consentire la riparazione del danno. Se il danno è troppo esteso e non può essere riparato, si può avere apoptosi Se il danno non viene riparato e la cellula non va incontro ad apoptosi, il danno del filamento di DNA induce una mutazione nel filamento figlio se la cellula si replica i livelli di mutazione aumentano con la velocità di divisione cellulare

66
Types and rates of mutation
Type Mechanism Frequency________ Genome chromosome per cell division mutation misaggregation (e.g., aneuploidy) Chromosome chromosome 6 X 10-4 per cell division mutation rearrangement (e.g., translocation) Gene base pair mutation per base pair per mutation (e.g., point mutation, cell division or or small deletion or per locus per insertion generation This slide shows the three basic types of mutational events and their frequencies. We will be concentrating on "gene mutations," which are base pair mutations or small deletions or insertions.
 Chromosome chromosome 6 X 10-4 per cell division. mutation rearrangement. (e.g., translocation) Gene base pair mutation per base pair per. mutation (e.g., point mutation, cell division or. or small deletion or per locus per. insertion generation. This slide shows the three basic types of mutational events and their frequencies. We will be concentrating. on gene mutations, which are base pair mutations or small deletions or insertions.")
67
Summary of DNA lesions Missing base Acid and heat depurination (~104 purines per day per cell in humans) Altered base Ionizing radiation; alkylating agents Incorrect base Spontaneous deaminations cytosine to uracil adenine to hypoxanthine Deletion-insertion Intercalating reagents (acridines) Dimer formation UV irradiation Strand breaks Ionizing radiation; chemicals (bleomycin) Interstrand cross-links Psoralen derivatives; mitomycin C (Tautomer formation Spontaneous and transient) As shown here, DNA is prone to many different kinds of damaging reactions, any of which can alter DNA function and cause mutations if not repaired.
 Dimer formation UV irradiation. Strand breaks Ionizing radiation; chemicals (bleomycin) Interstrand cross-links Psoralen derivatives; mitomycin C. (Tautomer formation Spontaneous and transient) As shown here, DNA is prone to many different kinds of damaging reactions, any of which can alter DNA. function and cause mutations if not repaired.")
68
Types of base pair mutations
normal sequence CATTCACCTGTACCA GTAAGTGGACATGGT base pair substitutions transition: pyrimidine to pyrimidine transversion: pyrimidine to purine transversion (T-A to G-C) transition (T-A to C-G) CATGCACCTGTACCA GTACGTGGACATGGT CATCCACCTGTACCA GTAGGTGGACATGGT This slide illustrates the four basic types of base pair mutations. Two of them result in the conversion of one base pair to another (base pair substitution). The others result in removal (deletion) or addition (insertion) of one or more base pairs. (Note that a transition mutation results when a pyrimidine on one strand is converted to another pyrimidine on the same strand. The complementary strand would see a conversion from one purine to the other purine.) deletion insertion CATCACCTGTACCA GTAGTGGACATGGT CATGTCACCTGTACCA GTACAGTGGACATGGT deletions and insertions can involve one or more base pairs
 transition (T-A to C-G) CATGCACCTGTACCA. GTACGTGGACATGGT. CATCCACCTGTACCA. GTAGGTGGACATGGT. This slide illustrates the four basic types of base pair mutations. Two of them result in the conversion of one. base pair to another (base pair substitution). The others result in removal (deletion) or addition (insertion) of. one or more base pairs. (Note that a transition mutation results when a pyrimidine on one strand is converted. to another pyrimidine on the same strand. The complementary strand would see a conversion from one purine. to the other purine.) deletion. insertion. CATCACCTGTACCA. GTAGTGGACATGGT. CATGTCACCTGTACCA. GTACAGTGGACATGGT. deletions and insertions can involve one. or more base pairs.")
69
Spontaneous mutations can be caused by tautomers
Tautomeric forms of the DNA bases Adenine There are many different causes of mutations. Spontaneous mutations (those that result from no external cause) can occur simply by rearrangement of bonds and by the repositioning of hydrogens in the purine and pyrimidine bases. The common forms of adenine and cytosine are the amino forms, which can rearrange to the imino forms. The repositioning of hydrogens changes their base pairing, hydrogen bonding chemistry. Cytosine AMINO IMINO
 can occur simply by rearrangement of bonds and by the repositioning of hydrogens in the purine and. pyrimidine bases. The common forms of adenine and cytosine are the amino forms, which can rearrange to. the imino forms. The repositioning of hydrogens changes their base pairing, hydrogen bonding chemistry. Cytosine. AMINO. IMINO.")
70
Tautomeric forms of the DNA bases
Guanine The common forms of guanine and thymine are the keto forms, which can rearrange to the enol forms. Thymine KETO ENOL

71
Mutation caused by tautomer of cytosine
Normal tautomeric form Guanine Cytosine This figure illustrates how a conversion from the amino to the imino form of cytosine changes the locations of hydrogen donor and acceptor groups, such that the imino form of cytosine base pairs with adenine instead of guanine. As shown in the next figure, this will ultimately lead to a transition mutation. As for the other tautomers, the imino form of adenine "looks sort of like" a guanine and should be able to form two hydrogen bonds with cytosine; the enol form of guanine looks like an adenine and should be able to form three hydrogen bonds with thymine; and the enol form of thymine looks like a cytosine and should be able to form three hydrogen bonds with guanine. In all cases, the wrong nucleotide will be inserted into the growing DNA chain. Rare imino tautomeric form Adenine cytosine mispairs with adenine resulting in a transition mutation

72
C G C G C G C G C A C G C A T A Mutation is perpetuated by replication
and replication of C-G should give daughter strands each with C-G C G C A and C G tautomer formation C during replication will result in mispairing and insertion of an improper A in one of the daughter strands The conversion of a C-G base pair to a T-A base pair takes two steps. If the tautomeric form of cytosine is present during DNA replication, an adenosine will be inserted into the daughter DNA strand (instead of the normal guanosine). During the next round of DNA replication, the adenosine then serves as a template for the insertion of a thymidine in the new DNA strand, resulting in a transition mutation (the conversion of a C-G to a T-A). C A T A which could result in a C-G to T-A transition mutation in the next round of replication, or if improperly repaired
. During the next round of DNA replication, the adenosine then serves as a template for the. insertion of a thymidine in the new DNA strand, resulting in a transition mutation (the conversion of a C-G to a. T-A). C. A. T. A. which could result in a C-G to T-A transition mutation in the next. round of replication, or if improperly repaired.")
73
Chemical mutagens Deamination by nitrous acid
Mutation can also occur by the action of chemical mutagens. This figure shows how oxidative deamination of cytosine converts it to uracil, and how the oxidative deamination of adenine converts it to hypoxanthine, both processes altering the hydrogen bonding specificities of these bases.

74
Derivation by hydroxylamine
Alkylation by dimethyl sulfate causes depurination This figure shows two more ways DNA can be chemically mutagenized. In the upper example, cytosine is shown being derivatized by hydroxylamine. Anything that alters the base pairing potential or the specificity of interation between a base and a DNA binding protein, can alter DNA function. In the lower example, guanine is shown being derivatized by dimethyl sulfate (DMS). This results in the release of the base from the phosphodiester backbone of DNA. The formation of a quarternary nitrogen destabilizes the deoxyriboside bond and the base is released from deoxyribose
. This results in the release of the base from the. phosphodiester backbone of DNA. The formation of a quarternary nitrogen destabilizes the. deoxyriboside bond and the base is released from deoxyribose.")
75
Attack by oxygen radicals
The deoxyribose ring is also susceptible to damage, here being shown being cleaved by an oxygen free radical. This would break the phosphodiester backbone of DNA.

76
Thymine dimer formation by UV light
Sunlight is particularly damaging to DNA. This figure shows the formation of a thymine dimer, catalyzed by UV light. The thymine dimer bridges two adjacent thymine residues on the same DNA strand.

77
DNA damage, repair mechanisms and consequences
DNA damage, repair mechanisms and consequences. a, Common DNA damaging agents (top); examples of DNA lesions induced by these agents (middle); and most relevant DNA repair mechanism responsible for the removal of the lesions (bottom). b, Acute effects of DNA damage on cell-cycle progression, leading to transient arrest in the G1, S, G2 and M phases (top), and on DNA metabolism (middle). Long-term consequences of DNA injury (bottom) include permanent changes in the DNA sequence (point mutations affecting single genes or chromosome aberrations which may involve multiple genes) and their biological effects. Abbreviations: cis-Pt and MMC, cisplatin and mitomycin C, respectively (both DNA-crosslinking agents); (6–4)PP and CPD, 6–4 photoproduct and cyclobutane pyrimidine dimer, respectively (both induced by UV light); BER and NER, base- and nucleotide-excision repair, respectively; HR, homologous recombination; EJ, end joining.
; examples of DNA lesions induced by these agents (middle); and most relevant DNA repair mechanism responsible for the removal of the lesions (bottom). b, Acute effects of DNA damage on cell-cycle progression, leading to transient arrest in the G1, S, G2 and M phases (top), and on DNA metabolism (middle). Long-term consequences of DNA injury (bottom) include permanent changes in the DNA sequence (point mutations affecting single genes or chromosome aberrations which may involve multiple genes) and their biological effects. Abbreviations: cis-Pt and MMC, cisplatin and mitomycin C, respectively (both DNA-crosslinking agents); (6–4)PP and CPD, 6–4 photoproduct and cyclobutane pyrimidine dimer, respectively (both induced by UV light); BER and NER, base- and nucleotide-excision repair, respectively; HR, homologous recombination; EJ, end joining.")
78
Excision repair (base or nucleotide)
Base excision repair Nucleotide excision repair ATGCUGCATTGA TACGGCGTAACT ATGC GCATTGA AT GCATTGA deamination ATGCCGCATTGA uracil DNA glycosylase repair nucleases DNA polymerase b DNA ligase thymine dimer ATGCUGCATTGATAG TACGGCGTAACTATC excinuclease AT AG TACGGCGTAACTATC (~30 nucleotides) DNA polymerase b ATGCCGCATTGATAG TACGGCGTAACTATC There are two types of excision repair: base excision repair (left) and nucleotide excision repair (right). While they differ in their initial steps (top), they are similar in the latter steps (bottom). If cytosine is deaminated forming uracil, the U can be recognized as being an improper base in DNA by the enzyme, uracil DNA glycosylase. This enzyme cleaves the uracil base from the phosphodiester backbone, and the space is opened up by repair nucleases that remove a number of nucleotides from one strand (the other strand has to be left intact to serve as the template for DNA repair). The repair polymerase, DNA polymerase beta, then fills in the gap and DNA ligase seals the last phosphodiester bond. The double strandedness of DNA makes possible both DNA replication and DNA repair, because the template strand always contains the information for the synthesis of a complementary strand. Nucleotide excision repair occurs when the DNA lesion is larger, for example when there is a thymine dimer. In this case, a special repair excinuclease removes about 30 nucleotides, including the lesion. The DNA is then resynthesized and ligated together as with base excision repair. DNA ligase ATGCCGCATTGATAG TACGGCGTAACTATC
 DNA polymerase b. ATGCCGCATTGATAG. TACGGCGTAACTATC. There are two types of excision repair: base excision repair (left) and nucleotide excision repair (right). While. they differ in their initial steps (top), they are similar in the latter steps (bottom). If cytosine is deaminated forming uracil, the U can be recognized as being an improper base in DNA by the. enzyme, uracil DNA glycosylase. This enzyme cleaves the uracil base from the phosphodiester backbone, and. the space is opened up by repair nucleases that remove a number of nucleotides from one strand (the other. strand has to be left intact to serve as the template for DNA repair). The repair polymerase, DNA polymerase. beta, then fills in the gap and DNA ligase seals the last phosphodiester bond. The double strandedness of DNA. makes possible both DNA replication and DNA repair, because the template strand always contains the. information for the synthesis of a complementary strand. Nucleotide excision repair occurs when the DNA lesion is larger, for example when there is a thymine dimer. In this case, a special repair excinuclease removes about 30 nucleotides, including the lesion. The DNA is then. resynthesized and ligated together as with base excision repair. DNA ligase. ATGCCGCATTGATAG. TACGGCGTAACTATC.")
79
Mechanism for base-excision repair

80
Homologous recombination: riparazione di rotture della doppia elica
the 5'–3' exonuclease activity exposes both 3' ends. Identification of a homologous sequence. After identification of the identical sister chromatid sequence, the intact double-stranded copy is used as a template to properly heal the broken ends by DNA synthesis (III). Finally, the so-called Holliday-junctions are resolved by resolvases
. Finally, the so-called Holliday-junctions are resolved by resolvases.")
81
Mismatch (post-replication) repair
Heterodimers of hMSH2/6 recognize single-base or insertion/deletion loops Heterodimeric complexes interact with MSH complexes and replication factors. Excision of the new strand past the mismatch and resynthesis N.B. la riparazione post-replicativa è error prone

82
Mechanisms of Repair Mutations that occur spontaneously any time are repaired by excision repair (base excision or nucleotide excision) Mutations that occur during DNA replication are repaired when possible by proofreading by the DNA polymerases Mutations that are not repaired by proofreading are repaired by mismatched (post-replication) repair followed by excision repair
 repair followed by. excision repair.")
83
Difetti congeniti dei meccanismi di riparazione del DNA predispongono allo sviluppo di tumori.

84
Defects in DNA repair or replication
All are associated with a high frequency of chromosome and gene (base pair) mutations; most are also associated with a predisposition to cancer, particularly leukemia Xeroderma pigmentosum caused by mutations in genes involved in nucleotide excision repair associated with a 2000-fold increase of sunlight-induced skin cancer and with other types of cancer such as melanoma Ataxia telangiectasia caused by gene that detects DNA damage increased risk of X-ray associated with increased breast cancer in carriers Fanconi anemia sensitivity to sunlight Bloom syndrome caused by mutations in a a DNA helicase gene Cockayne syndrome caused by a defect in transcription-linked DNA repair Werner’s syndrome caused by mutations in a DNA helicase gene premature aging As shown here, there are a number of defects of DNA replication and repair that are associated with a predisposition to cancer and other disorders. This highlights the importance of high fidelity DNA replication and for the presence of DNA repair mechanisms for normal cell function and longevity.
 mutations; most are also associated with a. predisposition to cancer, particularly leukemia. Xeroderma pigmentosum. caused by mutations in genes involved in nucleotide excision repair. associated with a 2000-fold increase of sunlight-induced. skin cancer and with other types of cancer such as melanoma. Ataxia telangiectasia. caused by gene that detects DNA damage. increased risk of X-ray. associated with increased breast cancer in carriers. Fanconi anemia. sensitivity to sunlight. Bloom syndrome. caused by mutations in a a DNA helicase gene. Cockayne syndrome. caused by a defect in transcription-linked DNA repair. Werner’s syndrome. caused by mutations in a DNA helicase gene. premature aging. As shown here, there are a number of defects of DNA replication and repair that are associated with a. predisposition to cancer and other disorders. This highlights the importance of high fidelity DNA replication. and for the presence of DNA repair mechanisms for normal cell function and longevity.")
85
human elephant cow Life span hamster rat mouse shrew
Correlation between DNA repair activity in fibroblast cells from various mammalian species and the life span of the organism There is a direct correlation between DNA repair enzymatic activity and the life span of organisms, suggesting that DNA repair activity slows down cellular senescence and that cellular senescence is caused by mutations in DNA. Defects in DNA repair or replication can lead to a number of abnormalities. 100 human elephant cow Life span 10 There is a direct correlation between DNA repair enzymatic activity and the life span of organisms, suggesting that DNA repair activity slows down cellular senescence and that cellular senescence is caused by mutations in DNA. Defects in DNA repair or replication can lead to a number of abnormalities. hamster rat mouse shrew 1 DNA repair activity

86
Se i meccanismi di riparazione del DNA falliscono e la cellula non si attivano i meccanismi dell’apoptosi, la mutazione viene fissata nelle cellule figlie. Se la mutazione altera la funzionalità di geni che stimolano (proto-oncogeni) o inibiscono la progressione della cellula nel ciclo cellulare (geni repressori dei tumori; p53 induce anche l’apoptosi), si ha la trasformazione neoplastica della cellula.
 o inibiscono la progressione della cellula nel ciclo cellulare (geni repressori dei tumori; p53 induce anche l’apoptosi), si ha la trasformazione neoplastica della cellula.")
88
S G0 G1 G2 M The role of p53 in the cell cycle p53
apoptosis (cell death) DNA synthesis UV irradiation leads to cell cycle arrest p53 S phase G0 G1 phase Quiescent cells G2 The p53 gene functions in the normal cell as part of a mechanism that senses when the cellular DNA has been damaged, for example by UV irradiation. As described in the previous slide, p53 causes cell cycle arrest to allow the cell to repair the damaged DNA, or if the cell is too damaged, will cause the cell to enter an apoptotic pathway. phase M Growth and preparation for cell division phase Mitosis
 DNA synthesis. UV irradiation leads. to cell cycle arrest. p53. S. phase. G0. G1. phase. Quiescent cells. G2. The p53 gene functions in the normal cell as part of a mechanism that senses when the cellular DNA has been damaged, for example by UV irradiation. As described in the previous slide, p53 causes cell cycle arrest to allow the cell to repair the damaged DNA, or if the cell is too damaged, will cause the cell to enter an apoptotic pathway. phase. M. Growth and. preparation for. cell division. phase. Mitosis.")
89
Multistep carcinogenesis
Stages in the evolution of colon cancer Chromosome 5q gene loss or mutation Normal colon cell Increased cell growth Ras gene mutation Adenoma I Chromosome 18 loss or mutation DCC tumor suppressor gene Adenoma II Progression from a normal cell to a fully metastatic tumor requires mutations in a number of genes. As shown here for the evolution of metastatic colon carcinoma, there are seven identifiable steps. Mutational events known to occur at some of these steps are indicated. These involve a loss of several chromosome regions (or entire chromosomes) and at least one gene mutation involving the Ras oncogene. While there may be a number of different routes from normal to metastatic, this illustrates that this is a multistep process. Much of the rest of this lecture will discuss the types of genes that are mutated or lost during tumorigenesis. Adenoma III Carcinoma Chromosome 17 loss or mutation p53 tumor suppressor gene Metastasis Other chromosome losses
 and at least one gene mutation involving the Ras oncogene. While there may be a number of different routes from normal to metastatic, this illustrates that this is a multistep process. Much of the rest of this lecture will discuss the types of genes that are mutated or lost during tumorigenesis. Adenoma. III. Carcinoma. Chromosome 17 loss or mutation. p53 tumor suppressor gene. Metastasis. Other chromosome losses.")
90
Riparazione tissutale
Morte delle cellule danneggiate Sostituzione delle cellule morte (proliferazione) e della matrice extracellulare
 e della matrice extracellulare.")
91
Morte cellulare Livello di esposizione all’agente necrogenico: basso apoptosi; indotta da danno a DNA, anche indiretto, se la riparazione del DNA fallisce alto necrosi L’apoptosi elimina le cellule danneggiate senza reazione infiammatoria; previene inoltre la trasformazione neoplastica.

92
Proliferazione cellulare
Sono coinvolti vari tipi cellulari Negli organi parenchimali (fegato, rene, polmoni), il danno necrotico induce la produzione, da parte di cellule non parenchimali (macrofagi, cellule endoteliali), di fattori che stimolano la divisione cellulare e la sintesi di matrice extracellulare.
, il danno necrotico induce la produzione, da parte di cellule non parenchimali (macrofagi, cellule endoteliali), di fattori che stimolano la divisione cellulare e la sintesi di matrice extracellulare.")
93
S G0 G1 G2 M The mammalian cell cycle de-differenziazione
DNA synthesis and histone synthesis Rapid growth and preparation for DNA synthesis S phase G0 G1 phase Quiescent cells G2 phase The S (for synthesis) phase of the cell cycle is when chromosomes are replicated. This requires DNA synthesis and histone synthesis (to make the proteins that will package the newly replicated DNA). M Growth and preparation for cell division phase de-differenziazione Mitosis
 phase of the cell cycle is when chromosomes are replicated. This requires DNA. synthesis and histone synthesis (to make the proteins that will package the newly replicated DNA). M. Growth and. preparation for. cell division. phase. de-differenziazione. Mitosis.")
95
Il grado di divisione cellulare può essere valutato dalla sintesi di DNA l’incorporazione di 3H-timidina è un indicatore di sintesi di DNA.

96
Riparazione tissutale e necrosi
La necrosi di un tessuto, indotta da una sostanza citotossica, avviene quando l’entità del danno è tale da sormontare i meccanismi di riparazione: riparazione molecolare apoptosi sostituzione delle cellule danneggiate Ad es., il clordecone (insetticida), blocca la proliferazione cellulare in risposta a CCl4 CCl4 causa necrosi a dosi normalmente non tossiche.
, blocca la proliferazione cellulare in risposta a CCl4 CCl4 causa necrosi a dosi normalmente non tossiche.")
97
Effetti istologici di CCl4 nel fegato
Figure 2. Detection of PrP mRNA expression in rat liver tissue. Untreated ((a, c) ) and CCl4-treated ((b, d–f)) rat livers were stained with Azan-Mallory stain ((a, b) ) and in situ hybridization for PrP mRNA expression with antisense riboprobe ((c, d, f)) and sense probe ((e)).
 ) and CCl4-treated ((b, d–f)) rat livers were stained with Azan-Mallory stain ((a, b) ) and in situ hybridization for PrP mRNA expression with antisense riboprobe ((c, d, f)) and sense probe ((e)).")
98
Infiammazione Il danno tissutale causa il rilascio di citochine infiammatorie (TNF, IL-1) da parte dei macrofagi residenti (nel fegato le cellule di Kupffer) inizio della reazione infiammatoria. I mediatori dell’infiammazione aumentano il danno.
 da parte dei macrofagi residenti (nel fegato le cellule di Kupffer) inizio della reazione infiammatoria. I mediatori dell’infiammazione aumentano il danno.")
100
N.B.: GdCl3 elimina selettivamente le cellule di Kupffer
Institution: Corso di Laurea in Servizio Sociale - Univ. La Sapienza Sign In as Personal Subscriber N.B.: GdCl3 elimina selettivamente le cellule di Kupffer FIG. 1. GdCl3 protects against MCT/LPS-induced liver injury. LPS (7.4 x 106 EU/kg) or saline vehicle (Veh) was administered, iv, to rats 4 h after ip administration of MCT (100 mg/kg) or saline vehicle. Rats were pretreated with 10 mg GdCl3-6H2O/kg or saline vehicle, iv, 24 h before LPS administration. TNF- concentration (A), ALT (B) and AST (C) activities, and HA concentration (D) were evaluated in plasma 18 h after MCT administration.
 or saline vehicle (Veh) was administered, iv, to rats 4 h after ip administration of MCT (100 mg/kg) or saline vehicle. Rats were pretreated with 10 mg GdCl3-6H2O/kg or saline vehicle, iv, 24 h before LPS administration. TNF- concentration (A), ALT (B) and AST (C) activities, and HA concentration (D) were evaluated in plasma 18 h after MCT administration.")
101
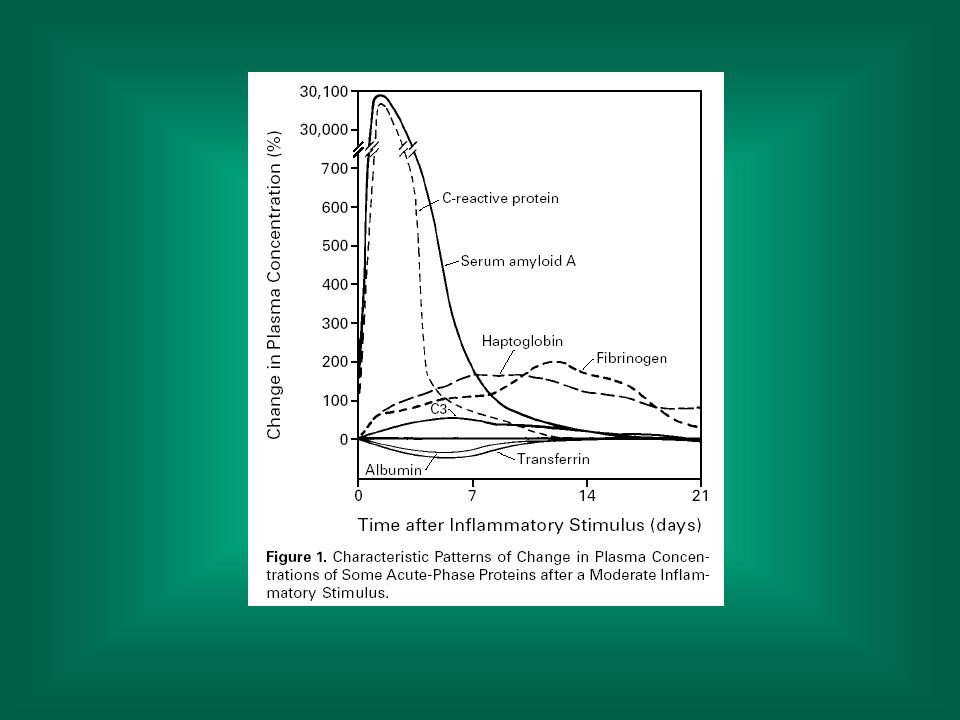
Le citochine rilasciate da macrofagi e cellule endoteliali alterano la sintesi delle proteine epatiche: proteine di fase acuta (positive): proteina C-reattiva, alfa2-macroglobulina, alfa1-antiproteasi ecc.; oltre a svolgere uno ruolo fisiologico (inibizione proteasi ecc.) hanno valore diagnostico. proteine di fase acuta negative: albumina, transferrina ecc.
: proteina C-reattiva, alfa2-macroglobulina, alfa1-antiproteasi ecc.; oltre a svolgere uno ruolo fisiologico (inibizione proteasi ecc.) hanno valore diagnostico. proteine di fase acuta negative: albumina, transferrina ecc.")
103
Fibrosi Danno cellulare proliferazione cellulare + produzione di matrice extracellulare, mediata principalmente da TGF-. La sovrapproduzione di TGF- cessa quando il danno tissutale è riparato. Se ciò non avviene, si sviluppa fibrosi.

104
Ratti trattati con CCl4 (s.c.) per 9 settimane
Fibrosi misurata come contenuto in idrossiprolina

105
The protein collagen is unusual in its widespread modification of proline to 4-hydroxyproline (also called hydroxyproline). Enzymatic hydroxylation of procollagen proline residues in the synthesis of collagen

Presentazioni simili