Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
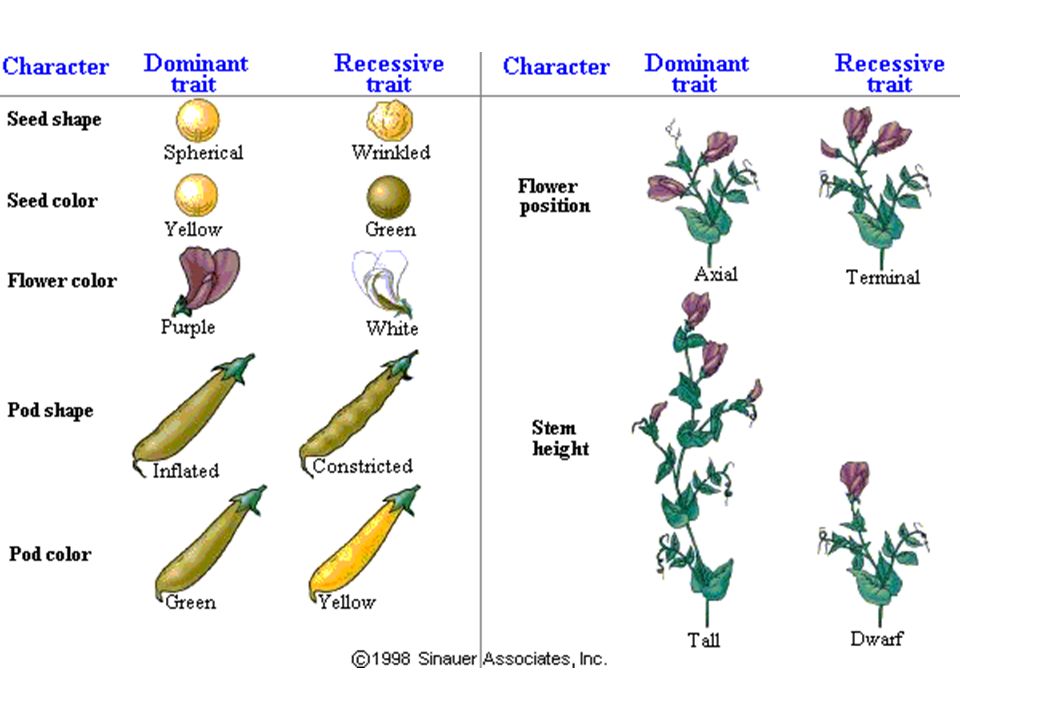
MENDEL Prima legge di Mendel AA aa A a Aa A a A AA Aa a Aa aa
Studio di “caratteri” che si presentano in forme alternative Prima legge di Mendel I fattori responsabili della trasmissione ereditaria di particolari tratti (geni) sono unità discrete che sono presenti in doppia copia nei genitori (alleli) e si separano (segregano) durante la formazione dei gameti. I gameti si combinano casualmente dando origine a 4 combinazioni alleliche nella F2 Il tratto “liscio” è dominante su “rugoso” (recessivo) Il tratto recessivo riappare nella generazione F2 in rapporto 3:1 AA aa Genitori A a Gameti Aa F1 A a A AA Aa F2 a Aa aa
 sono unità discrete che sono presenti in doppia copia nei genitori (alleli) e si separano (segregano) durante la formazione dei gameti. I gameti si combinano casualmente dando origine a 4 combinazioni alleliche nella F2. Il tratto liscio è dominante su rugoso (recessivo) Il tratto recessivo riappare nella generazione F2 in rapporto 3:1. AA. aa. Genitori. A. a. Gameti. Aa. F1. A. a. A. AA. Aa. F2. a. Aa. aa.")
3
INROCIO MONOIBRIDO P: lisci x rugosi F1: tutti lisci F2: lisci e 1850 rugosi (1:3)
")
4
Seconda legge di Mendel:
Assortimento indipendente dei caratteri Comportamento di due caratteri : Segregazione indipendente dei due alleli di un carattere (R r) rispetto a quelli dell’altro (Y y) Formazione di 4 tipi di gameti in proporzioni uguali Incontro casuale dei gameti -> comparsa di 4 fenotipi nella F2 nel rapporto 9:3:3:1 Fenotipi parentali + ricombinanti NB si riferisce solo a caratteri relativi a geni localizzati su cromosomi diversi o geni sullo stesso cromosoma distanti fra loro
 rispetto a quelli dell’altro (Y y) Formazione di 4 tipi di gameti in proporzioni uguali. Incontro casuale dei gameti -> comparsa di 4 fenotipi nella F2 nel rapporto 9:3:3:1. Fenotipi parentali + ricombinanti. NB si riferisce solo a caratteri relativi a geni localizzati su cromosomi diversi o geni sullo stesso cromosoma distanti fra loro.")
5
P1: liscio/giallo x rugoso/verde
RRYY rryy F1: tutti lisci/gialli RrYy x RrYy F2

7
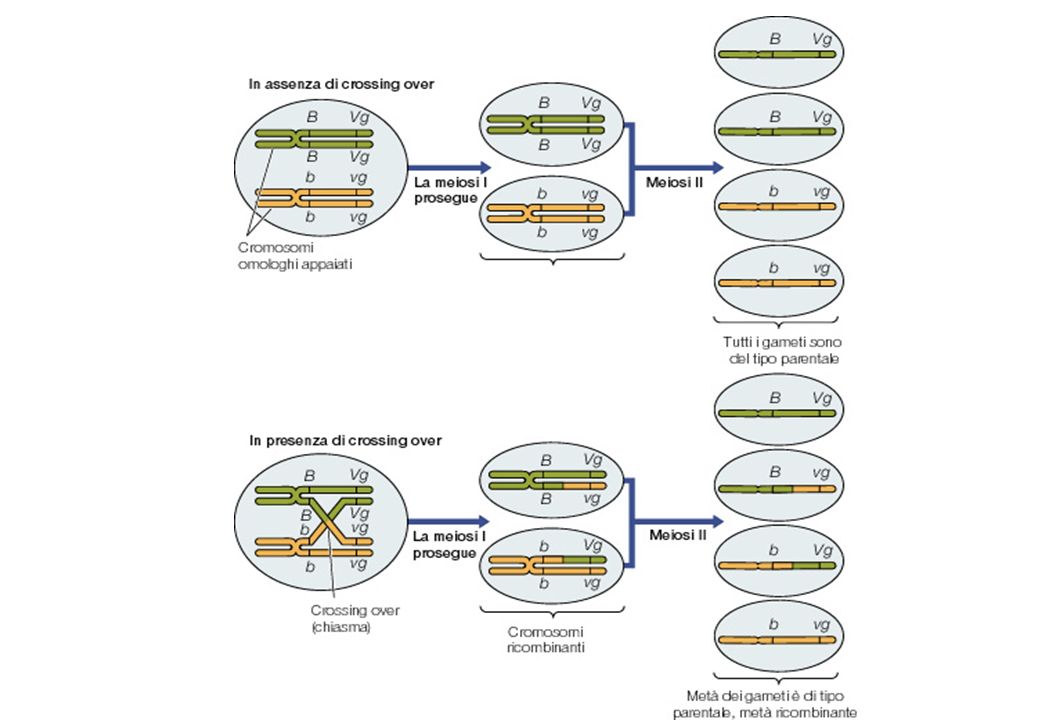
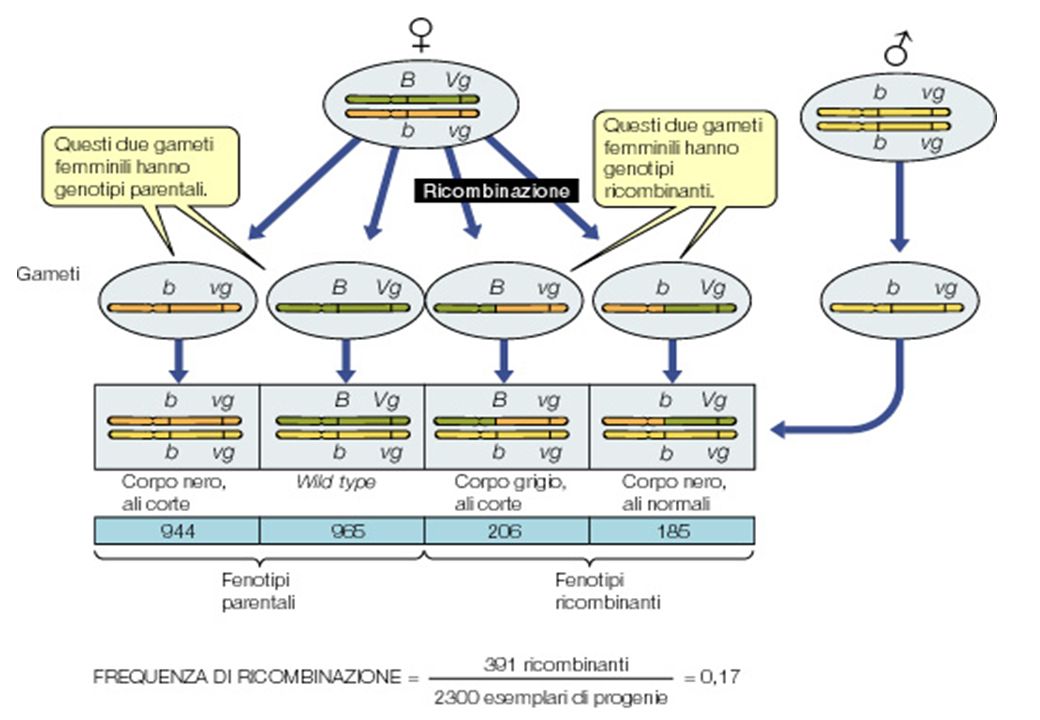
Morgan Studio della ricombinazione in Drosophila
BBVgVg x bbvgvg B-Vg b-vg b-Vg B-vg B b Vgvg b b vgvg b b Vgvg B b vgvg b-vg Morgan Studio della ricombinazione in Drosophila

10
Mappaggio di loci attraverso l’analisi della freq di ricombinazione

11
ANALISI DI LINKAGE DI CARATTERI MENDELIANI
Determinare la frequenza con cui due loci ricombinano fra loro alla meiosi = TETHA ( se due loci sono su cromosomi diversi o molto lontani sullo stesso cromosoma segregano indipendentemente. La probabilità che vengano ereditati insieme è del 1/2 -> = 50% Se due loci sono vicini fra loro sullo stesso cromosoma saranno ereditati insieme più frequentemente -> < 50% Tanto più sono vicini, tanto più piccola è la probabilità che avvenga un crossing-over LA FREQUENZA DI RICOMBINAZIONE () E’ UNA MISURA DELLA DISTANZA GENETICA
 E’ UNA MISURA DELLA DISTANZA GENETICA.")
12
DISTANZA GENETICA = numero atteso di crossing-overs fra 2 loci per meiosi L’unità di misura della distanza genetica è il Morgan 1 Morgan = 100 centiMorgan (cM) In media 1 cM corrisponde a circa 106 bp. Genoma umano ≈ 3x109 = 33 Morgan Le frequenze di ricombinazione fra loci non sono additive (a causa dell’effetto dei crossing-over multipli) Per <10% c’è una corrispondenza ≈ 1:1 fra e cM : 1 cM ≈ = 1% Per <10% si usano delle funzioni di mappa per convertire in cM e viceversa Funzioni di mappa -> relazione fra frazione di ricombinazione e distanza genetica Haldane: = -1/2 ln(1-2) e-2) Kosambi tiene conto dell’interferenza A1 A2 B1 B2 C2 C1
 In media 1 cM corrisponde a circa 106 bp. Genoma umano ≈ 3x109 = 33 Morgan. Le frequenze di ricombinazione fra loci non sono additive (a causa dell’effetto dei crossing-over multipli) Per <10% c’è una corrispondenza ≈ 1:1 fra e cM : 1 cM ≈ = 1% Per <10% si usano delle funzioni di mappa per convertire in cM e viceversa. Funzioni di mappa -> relazione fra frazione di ricombinazione e distanza genetica. Haldane: = -1/2 ln(1-2) e-2) Kosambi tiene conto dell’interferenza. A1. A2. B1. B2. C2. C1.")
13
Alleli e genotipi Eterozigosità (H)
Ogni individuo ha due alleli ad ogni locus : genotipo Omozigote/Eterozigote Eterozigosità (H) Probabilità che un individuo preso a caso sia eterozigote per un determinato marker H = 1 - (p12 + p22 + p32 + p42 + …..pi2)
 Probabilità che un individuo preso a caso sia eterozigote per un determinato marker. H = 1 - (p12 + p22 + p32 + p42 + …..pi2)")
14
EQUILIBRIO DI HARDY-WEINBERG
Relazione tra frequenze alleliche e genotipiche Presupposto: nella popolazione le unioni sono casuali e non ci sono eventi di mutazione, selezione, migrazione in atto A a LOCUS CON 2 ALLELI A frequenza p a frequenza q p + q = 1 AA p2 Aa p q A a Aa p q a a q2 LOCUS AUTOSOMICO f(AA) = p2 f(Aa) = 2pq f(aa) = q2 p2 + 2pq + q2 = 1 LOCUS LEGATO AL CROM X Nei maschi: Nelle femmine f(A) = p f(a) = q f(AA) = p2 f(Aa) = 2pq f(aa) = q2
 = p2 f(Aa) = 2pq f(aa) = q2 p2 + 2pq + q2 = 1. LOCUS LEGATO AL CROM X. Nei maschi: Nelle femmine. f(A) = p f(a) = q f(AA) = p2 f(Aa) = 2pq f(aa) = q2.")
15
Genotipi, fenotipi e penetranza
Il fenotipo e’ la caratteristica che deriva dall’avere un determinato genotipo La probabilità che un determinato genotipo risulti nell’espressione di un fenotipo è la penetranza In certi casi la stesso fenotipo può essere determinato da diversi genotipi (fenocopie) Lo stesso genotipo può avere diverse manifestazioni fenotipiche (pleiotropia)
 Lo stesso genotipo può avere diverse manifestazioni fenotipiche (pleiotropia)")
16
ANALISI DI LINKAGE (CONCATENAZIONE)
L’analisi di linkage permette determinare la posizione cromosomica di un locus responsabile di una determinata malattia/carattere genetico rispetto a marcatori polimorfici la cui localizzazione è nota L’analisi di linkage è un approccio molto utile per il mappaggio e l’identificazione di geni responsabili di malattie genetiche Mendeliane (oltre 1,200 geni identificati) Più difficoltosa è l’identificazione dei numerosi geni implicati in malattie più comuni, ad ereditarietà complessa (pochissimi geni identificati finora)
 Più difficoltosa è l’identificazione dei numerosi geni implicati in malattie più comuni, ad ereditarietà complessa. (pochissimi geni identificati finora)")
17
POSITIONAL CLONING

18
per l’ANALISI DI LINKAGE DI CARATTERI MENDELIANI
REQUISITI per l’ANALISI DI LINKAGE DI CARATTERI MENDELIANI Una o più famiglie in cui segrega il carattere/malattia in questione 2) MARCATORI GENETICI Altamente polimorfici . Facilmente tipizzabili e stabili di generazione in generazione Posizione nota nel genoma
 MARCATORI GENETICI. Altamente polimorfici . Facilmente tipizzabili e stabili di generazione in generazione. Posizione nota nel genoma.")
19
MARCATORI GENETICI Marcatori polimorfici -> presenza di 2 o + alleli alternativi Generalmente non funzionali Facilmente tipizzabili e stabili di generazione in generazione Posizione nota nel genoma -> costruzione di Framework Maps Per tutti i cromosomi sono state create mappe genetiche di marcatori polimorfici

20
MARCATORI MOLECOLARI (DEL DNA)
RFLPs (Restriction Fragment Length Polymorphisms) Presenza/assenza sito di taglio di enzima di restrizione Biallelici Southern blot / PCR MICROSATELLITI ripetiz in tandem di nucleotidi (CA)n Molti alleli, molto informativi Distribuiti in modo uniforme nel genoma. ≈ ogni 100 Kb PCR / marcatura con fluorescenza SNPs (Single Nucleotyde Polymorphisms) Differenze di singola base, non necessariamente riconosciuti da enz. di restriz. Molti frequenti Sviluppo di tecniche di genotyping automatizzate e in larga scala
 Presenza/assenza sito di taglio di enzima di restrizione. Biallelici. Southern blot / PCR. MICROSATELLITI. ripetiz in tandem di nucleotidi (CA)n. Molti alleli, molto informativi. Distribuiti in modo uniforme nel genoma. ≈ ogni 100 Kb. PCR / marcatura con fluorescenza. SNPs (Single Nucleotyde Polymorphisms) Differenze di singola base, non necessariamente riconosciuti da enz. di restriz. Molti frequenti. Sviluppo di tecniche di genotyping automatizzate e in larga scala.")
21
ANALISI GENETICA DI CARATTERI UMANI
Impossibilità di eseguire incroci “sperimentali” Famiglie piccole e tempi di generazione lunghi Alberi genealogici I 1 2 II 1 2 3 4 5 III 1 2 3 4 5 6 7 IV 1 2 3 4 5 V 1 2

22
Tratto AUTOSOMICO DOMINANTE
Una persona affetta ha almeno un genitore affetto I due sessi sono colpiti in modo uguale Carattere trasmesso da ambedue i sessi Un figlio di un affetto x non-affetto ha il 50% di probabilità di essere affetto (se genitore affetto=eterozigote) La > parte delle mutazioni che causano malattie dominanti (rare) sono presenti solo in eterozigosi Aa aa
 La > parte delle mutazioni che causano malattie dominanti (rare) sono presenti solo in eterozigosi. Aa. aa.")
23
Tratto AUTOSOMICO RECESSIVO
Di solito i genitori di individui affetti sono sani (portatori asintomatici) I due sessi sono colpiti in modo uguale Se due genitori sono portatori la probabilità di avere un figlio affetto è 1/4 Maggiore frequenza di consanguineità aa Aa A a A a AA
 I due sessi sono colpiti in modo uguale. Se due genitori sono portatori la probabilità di avere un figlio affetto è 1/4. Maggiore frequenza di consanguineità. aa. Aa. A a. A. a. AA.")
24
Cromosomi X e Y -> eteromorfi
Crom X contiene migliaia di geni Crom Y costituito per lo piu’ da eterocromatina, pochi geni funzionali SRY -> gene determinante il sesso maschile Regioni pseudoautosomiche (PAR): regioni con omologia al cromosoma X Sono le regioni dove ha luogo l’appaiamento e il crossing over col crom X Il crossing over a livello delle PAR assicura la corretta segregazione X-Y PAR1 -> crossing over obbligato -> Altissima freq di ricombinazione SRY
: regioni con omologia al cromosoma X. Sono le regioni dove ha luogo l’appaiamento e il crossing over col crom X. Il crossing over a livello delle PAR assicura la corretta segregazione X-Y. PAR1 -> crossing over obbligato -> Altissima freq di ricombinazione. SRY.")
25
INATTIVAZIONE DEL CROMOSOMA X
NB inattivazione non random nei tessuti extra-embrionali

26
Tratto RECESSIVO LEGATO AL CROMOSOMA X
X x XX Xx X Y XY xY xY Xx XY X X xX xX x Y xX xY XY XY Gli individui affetti sono prevalentemente maschi (emizigoti) I genitori di maschi affetti sono normali: la madre è una portatrice asintomatica Le femmine possono essere affette se la madre portatrice ed il padre è affetto. -> Talvolta come conseguenza del pattern di inattivazione del cromosoma X Il carattere non puo’ essere trasmesso da padre a figlio
 I genitori di maschi affetti sono normali: la madre è una portatrice asintomatica. Le femmine possono essere affette se la madre portatrice ed il padre è affetto. -> Talvolta come conseguenza del pattern di inattivazione del cromosoma X. Il carattere non puo’ essere trasmesso da padre a figlio.")
27
Tratto DOMINANTE LEGATO AL CROMOSOMA X
XY xx xY Xx xY xY Xx Xx Xx Xx XY xx Xx xY Ambedue i sessi sono affetti, ma femmine > maschi Spesso le femmine hanno sintomi più lievi o più variabili dei maschi Se un maschio è affetto, trasmetterà il carattere a tutte le figlie e a nessun figlio Se una femmina è affetta, trasmetterà il carattere alla metà dei figli, maschi o femmine

28
Metodo Parametrico (o Model based)
ANALISI DI LINKAGE Metodo Parametrico (o Model based) Definizione di un modello di ereditarietà del locus-malattia gene malattia bi-allelico con frequenze alleliche e penetranze conosciute Esempio: Singolo locus D = allele “mutato” d = allele wild-type Frequenze alleliche: P(D) = p, P(d) = 1- p = q Penetranze: fDD , fDd , fdd La posizione del gene malattia è considerato sconosciuto -> Stima di fra il locus malattia ed il marcatore Conteggio dei ricombinanti = ___R___ (R + NR) Si stima la posizione genetica del locus malattia calcolando la likelihood dei dati osservati al variare della frazione di ricombianzione
 Definizione di un modello di ereditarietà del locus-malattia. gene malattia bi-allelico con frequenze alleliche e penetranze conosciute. Esempio: Singolo locus D = allele mutato d = allele wild-type. Frequenze alleliche: P(D) = p, P(d) = 1- p = q. Penetranze: fDD , fDd , fdd. La posizione del gene malattia è considerato sconosciuto. -> Stima di fra il locus malattia ed il marcatore. Conteggio dei ricombinanti = ___R___. (R + NR) Si stima la posizione genetica del locus malattia calcolando la likelihood dei dati osservati al variare della frazione di ricombianzione.")
29
Metodo del LOD SCORE (Metodo della massima verosimiglianza) Confronto della probabilità dei genotipi osservati nelle ipotesi alternative di linkage o assortimento indipendente Ho = i due loci sono indipendenti -> = 0.5 H1 = i due loci sono in linkage -> 0 < < 0.5 Likelihood [L()] = Probabilità che i dati osservati si verifichino quando la fraz di ricombinaz è Likelihood ratio test: è significativamente < 0.5? LOD = log of the odds Z() = log10 [L()/L( =0.5)] Calcolo di pedigree likelihood: Algoritmi Elston-Stewart, Lander-Green, MCMC
] = Probabilità che i dati osservati si verifichino quando la fraz di ricombinaz è Likelihood ratio test: è significativamente < 0.5 LOD = log of the odds Z() = log10 [L()/L( =0.5)] Calcolo di pedigree likelihood: Algoritmi Elston-Stewart, Lander-Green, MCMC.")
30
LOD SCORE (LOD) Z = log10 R ( 1- ) NR 0.5 (R+NR) Per = 0
ESEMPIO CONTEGGIO RICOMBINANTI E CALCOLO DEL LOD SCORE: 1) Pedigree a fase nota 1)Modello di ereditarietà: Deduco i genotipi per locus malattia d d D d 2 5 1 1 1 2 3 4 2)LA FASE E’ NOTA 3)Individuo i ricombinanti NR NR NR NR NR R LOD SCORE (LOD) Per = 0 No ricomb Z = log 0.5 N > 1 ricomb. Z = - ∞ Z = log R ( 1- ) NR 0.5 (R+NR)
 Pedigree a fase nota. 1)Modello di ereditarietà: Deduco i genotipi per locus malattia. d d. D d )LA FASE E’ NOTA. 3)Individuo i ricombinanti. NR NR NR NR NR R. LOD SCORE (LOD) Per = 0. No ricomb Z = log N. > 1 ricomb. Z = - ∞ Z = log10 R ( 1- ) NR. 0.5 (R+NR)")
31
Z = log10 R ( 1- ) NR + NR ( 1- ) R 0.5 R+NR LOD SCORE (LOD)
ESEMPIO CONTEGGIO RICOMBINANTI E CALCOLO DEL LOD SCORE: 2) Pedigree a fase non nota 1)Modello di ereditarietà: Deduco i genotipi per locus malattia 2)LA FASE NON E’ NOTA ? 3)Individuo i ricombinanti D d D d 1 2 d d 2 1 3 4 D d D d d d D d d d D d NR NR NR NR NR R Se la fase è D d 1 2 R R R R R NR 2 1 Z = log R ( 1- ) NR NR ( 1- ) R 0.5 R+NR LOD SCORE (LOD) 1 2 1 2
 Pedigree a fase non nota. 1)Modello di ereditarietà: Deduco i genotipi per locus malattia. 2)LA FASE NON E’ NOTA. 3)Individuo i ricombinanti. D d. D d d d D d. D d. d d. D d. d d. D d NR NR NR NR NR R. Se la fase è. D d R R R R R NR Z = log10 R ( 1- ) NR + NR ( 1- ) R. 0.5 R+NR. LOD SCORE (LOD)")
32
Massimo Lod score (MLS)
Spesso i dati ricavati da una solo famiglia non sono sufficienti per stabilire presenza /assenza di linkage. I lod score ottenuti da famiglie indipendenti (per lo stesso valore di ) si possono sommare fra loro Si calcola il lod score per diversi valori di ( = 0, 0.1, 0.2,…, 0,5) -> Tabella Massimo Lod score (MLS) Valore max di lod score che si ottiene al variare di un parametro (generalmente ) Il valore di per cui il LOD è massimo è la stima più probabile della frazione di ricombinazione tetha 0.1 0.2 0.3 0.4 Fam 1 -inf 0.28 0.32 0.22 0.08 Fam2 1.20 1.02 0.82 0.58 Fam3 1.36 2.68 2.71 1.91 Totale 2.66 3.82 3.51 2.31
 si possono sommare fra loro. Si calcola il lod score per diversi valori di ( = 0, 0.1, 0.2,…, 0,5) -> Tabella. Massimo Lod score (MLS) Valore max di lod score che si ottiene al variare di un parametro (generalmente ) Il valore di per cui il LOD è massimo è la stima più probabile della frazione di ricombinazione. tetha Fam 1. -inf Fam Fam Totale")
33
linked, no recombination

34
ESEMPIO DI CALCOLO LOD SCORE
2 2 1 2 1 2 3 4 1 2 2 2 NR NR NR R NR NR NR NR NR R R R R Z = log ( 1- ) 4 0.5 5 Z = log /2 ( 1- ) /2 4 0.5 4
 Z = log10 1/2 ( 1- ) 4 + 1/2 ")
35
Costruzione di mappe genetiche di riferimento
FAMIGLIE CEPH Costituiscono un pannello di 40 famiglie di riferimento, selezionate per la loro struttura ideale per l’analisi di linkage (3 generazioni, con i 4 nonni, 2 genitori, e almeno 6 figli). Mappe genetiche dell’intero genoma: Généthon Marshfield deCODE Passo fondamentale per: Mappaggio di loci di malattie Hanno costituito una base per la costruzione e la verifica di mappe fisiche
. Mappe genetiche dell’intero genoma: Généthon Marshfield deCODE Passo fondamentale per: Mappaggio di loci di malattie. Hanno costituito una base per la costruzione e la verifica di mappe fisiche.")
37
Corrispondenza distanza genetica - fisica
Approssimativamente: 1 cM ~ 1 Mb (1x106 bp) Non è costante Varia da regione a regione (> verso i telomeri) Mappa femminile è più lunga Hotspots
 Non è costante. Varia da regione a regione (> verso i telomeri) Mappa femminile è più lunga. Hotspots.")
38
TABELLA LOD SCORES A 2 PUNTI
(Abifadel et al. Nat Genet 34:154, 2003)
")
39
MULTIPOINT LOD SCORE Data una mappa di markers con posizione nota, si calcola la likelihood per ogni posizione del locus malattia lungo il cromosoma. Permette di estrarre il massimo dell’informazione data da tutti i markers sul cromosoma. MLS = 4.80

40
Genome screen Limiti di significatività
Uso una collezione di marcatori uniformemente spaziati su tutto il genoma Tipicamente ~ 400 microsatelliti (10 cM distanza media) Calcolo Lod Score (multipoint) lungo tutti i cromosomi -> Test multipli Limiti di significatività Z > 3 evidenza di linkage è significativa E’ stato calcolato che questo corrisponde ad un “whole-genome p value” di 0.05: un falso positivo ogni 20 genome screens Z < -2 si puo’ escludere la presenza di linkage
 Calcolo Lod Score (multipoint) lungo tutti i cromosomi. -> Test multipli. Limiti di significatività. Z > 3 evidenza di linkage è significativa. E’ stato calcolato che questo corrisponde ad un whole-genome p value di 0.05: un falso positivo ogni 20 genome screens. Z < -2 si puo’ escludere la presenza di linkage.")
41
COSTRUZIONE DI APLOTIPI: definizione della regione critica
Una serie di alleli per loci adiacenti che vengono ereditati insieme sullo stesso cromosoma formano un aplotipo

43
How to carry out a linkage study
Collect pedigree(s) several small nuclear pedigrees (parents and two or more affected children) for autosomal recessive traits
 several small nuclear pedigrees (parents and two or more affected children) for autosomal recessive traits.")
44
How to carry out a linkage study
Collect pedigree(s) several small nuclear pedigrees (parents and two or more affected children) for autosomal recessive traits for autosomal dominant traits, single extended pedigrees with several affected individuals in multiple generations are often large enough to find significant linkage
 several small nuclear pedigrees (parents and two or more affected children) for autosomal recessive traits. for autosomal dominant traits, single extended pedigrees with several affected individuals in multiple generations are often large enough to find significant linkage.")
45
How to carry out a linkage study
Collect pedigree(s) Is my disease: Autosomal or X-linked? Dominant or recessive? What is the disease allele frequency? and the disease phenotype penetrance? Are there phenocopies? Define, if possible, suitable genetic model for the disease
 Is my disease: Autosomal or X-linked Dominant or recessive What is the disease allele frequency and the disease phenotype penetrance Are there phenocopies Define, if possible, suitable genetic model for the disease.")
46
How to carry out a linkage study
Collect pedigree(s) What is the maximum lod-score that I can achieve in my family, or collection of families? What is the average lod-score I can expect to reach, if there is linkage? Define, if possible, suitable genetic model for the disease Evaluate power of study through simulation
 What is the maximum lod-score that I can achieve in my family, or collection of families What is the average lod-score I can expect to reach, if there is linkage Define, if possible, suitable genetic model for the disease. Evaluate power of study through simulation.")
47
How to carry out a linkage study
Collect pedigree(s) Define, if possible, suitable genetic model for the disease Evaluate power of study through simulation Type markers throughout the genome, or
 Define, if possible, suitable genetic model for the disease. Evaluate power of study through simulation. Type markers throughout the genome, or.")
48
How to carry out a linkage study
Collect pedigree(s) Define, if possible, suitable genetic model for the disease Evaluate power of study through simulation Type markers throughout the genome, or Type markers in or near candidate genes or regions
 Define, if possible, suitable genetic model for the disease. Evaluate power of study through simulation. Type markers throughout the genome, or. Type markers in or near candidate genes or regions.")
49
Results from linkage study
L O D - S C O R E T A B L E R E P O R T Marker D1S D1S D1S D1S D1S D1S D1S D1S D1S D1S D1S

50
S Chavanas et al: Localization of the Netherton Syndrome Gene to Chromosome 5q32, by Linkage Analysis and Homozygosity Mapping. Am J Hum Genet 66:914, 2000

51
Recombinants can be used to identify limits of disease locus critical region
(pedigree adapted from Vitale et al, 2001) 6 6 5 10 11 11 8 7 4 5 9 5 7 6 6 8 8 9 4 5 5 6 5 6 5 6 6 5 6 6 6 6 10 8 5 7 11 11 10 11 11 12 11 10 10 10 4 4 8 4 7 5 4 8 8 8 5 5 5 5 9 9 5 5 5 5 7 6 7 6 6 4 6 7 7 7 8 9 8 9 8 8 9 8 8 8 6 5 4 5 5 5 5 4 4 4 6 6 5 10 8 7 4 6 6 5 6 5 6 6 11 10 10 10 11 10 11 12 10 4 8 8 8 4 8 4 5 8 5 5 5 5 5 5 5 9 5 7 7 7 7 6 7 6 4 7 8 8 8 8 9 8 9 8 8 6 4 4 4 5 4 5 5 4
")
52
Complications in linkage analysis: Genetic heterogeneity
Genetic (or locus) heterogeneity is when multiple independent genes can cause the same disease phenotype Classic lod-score analysis assumes disease is caused by a single gene which is the same in all families For common conditions, heterogeneity is probably the rule rather than the exception Disease genes will be linked to markers located in different regions of the genome in different families Genetic heterogeneity can lead to false exclusion of linkage when linked and unlinked families are analyzed together
 heterogeneity is when multiple independent genes can cause the same disease phenotype. Classic lod-score analysis assumes disease is caused by a single gene which is the same in all families. For common conditions, heterogeneity is probably the rule rather than the exception. Disease genes will be linked to markers located in different regions of the genome in different families. Genetic heterogeneity can lead to false exclusion of linkage when linked and unlinked families are analyzed together.")
53
Complications in linkage analysis: Genetic heterogeneity
Carefully select families for homogeneous clinical features (e.g., age of onset in BRCA): sometimes there are no clear differences in phenotype Analyze single pedigrees large enough to produce significant lod-scores: may not always be available (e.g., recessive inheritance) results are confined to the given pedigree Perform linkage analysis allowing for heterogeneity
: sometimes there are no clear differences in phenotype. Analyze single pedigrees large enough to produce significant lod-scores: may not always be available (e.g., recessive inheritance) results are confined to the given pedigree. Perform linkage analysis allowing for heterogeneity.")
54
Complications in linkage analysis: Incomplete penetrance and phenocopies
Incomplete penetrance refers to absence of disease phenotype in individuals with disease genotype Non-penetrant carriers will appear as recombinants and may lead to false exclusion of linkage To avoid errors deriving from reduced penetrance, carry out an “affecteds-only” analysis A genetic model including reduced penetrance can be defined in lod-score analysis

55
Complications in linkage analysis: Incomplete penetrance and phenocopies
For common traits, non genetic cases (sometime called phenocopies) may co-exist with genetic ones in same family Intra-familial genetic heterogeneity may lead to the presence of affected individuals with different disease genotype in the same family As in the case of non-penetrant carriers, these will appear as recombinants and may lead to false exclusion of linkage Phenocopies can be accomodated in lod-score analysis by defining a non-zero penetrance for the normal disease genotype
 may co-exist with genetic ones in same family. Intra-familial genetic heterogeneity may lead to the presence of affected individuals with different disease genotype in the same family. As in the case of non-penetrant carriers, these will appear as recombinants and may lead to false exclusion of linkage. Phenocopies can be accomodated in lod-score analysis by defining a non-zero penetrance for the normal disease genotype.")
56
Complications in linkage analysis: Other possible sources of errors
Pedigree errors (e.g., non paternity) Misclassification of affected and unaffected individuals (errors in diagnosis) Misclassification of marker genotypes (typing errors) Incorrect marker map (in multipoint analysis) Incorrect estimates of marker allele frequencies in pedigrees with untyped individuals (especially in two point analysis)
 Misclassification of affected and unaffected individuals (errors in diagnosis) Misclassification of marker genotypes (typing errors) Incorrect marker map (in multipoint analysis) Incorrect estimates of marker allele frequencies in pedigrees with untyped individuals (especially in two point analysis)")
57
Marker allele frequencies can affect results of linkage analysis
Single nucleotide polymorphism (SNP) with alleles 1 and 2 2 2 If mother is 11, daughter is a recombinant Mother can be 11 or 12 1 2 If mother is 12, phase of daugher is unknown and chances are 50:50 that she is a recombinant or a non recombinant Probability of mother’s genotype depends on marker allele frequencies in the general population
 with alleles 1 and If mother is 11, daughter is a recombinant. Mother can be 11 or If mother is 12, phase of daugher is unknown and chances are 50:50 that she is a recombinant or a non recombinant Probability of mother’s genotype depends on marker allele frequencies in the general population.")
58
Complications in linkage analysis: Complex mode of inheritance
Many common traits result from interaction of several genes and environmental factors Disease transmission does not follow a simple Mendelian mode of inheritance Lod-score analysis assumes knowledge of genetic model underlying transmission of disease It may not be the best method for detection of genes responsible for complex traits

59
Programmi: http://linkage.rockefeller.edu/soft/list.html
Linkage, Fastlink, Vitesse, Simwalk, Merlin, Genehunter

Presentazioni simili






 Dominanza incompleta>")


 è dominante sul pelo liscio ( r )>")






>")





