Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Fibrosi Cistica

2
Malattia ereditaria monofattoriale
autosomica recessiva causata dalla presenza di una o più mutazioni nel gene che codifica per la CFTR localizzato sul cromosoma 7

3
Epidemiologia Nati/anno: oltre 200
Affetti: circa (60% bambini) Portatori: oltre Sopravvivenza media: 30 anni
 Portatori: oltre Sopravvivenza media: 30 anni.")
4
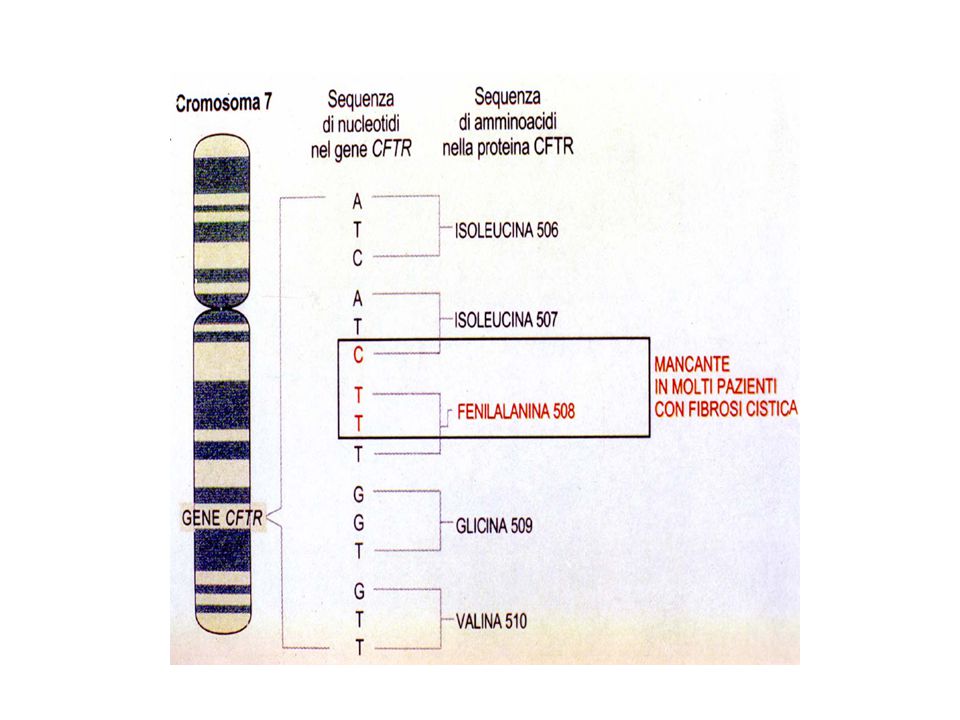
Gene della CFTR Composto da 230 kb
27 esoni che codificano per un mRNA maturo di 6129 aminoacidi Proteina 1480 aminoacidi

5
Mutazioni Numerose mutazioni più di 2000
Mutazioni puntiformi: missense, frameshift, delezioni ed inserzioni Le varie mutazioni hanno frequenza diversa La più frequente è la delta F508 (delezione di tre nucleotidi)
")
7
Classificazione delle mutazioni del gene della Fibrosi Cistica
Le mutazioni della fibrosi cistica (gene CFTR) sono state suddivise in 5 classi in baseal loro effetto funzionale (Modificato da Zielenski e Tsui, Ann Rev Genetics 29: 796, 1995).
 sono state suddivise in 5 classi in baseal loro effetto funzionale (Modificato da Zielenski e Tsui, Ann Rev Genetics 29: 796, 1995).")
8
Il CFTR è una proteina transmembrana con funzioni di canale ionico

9
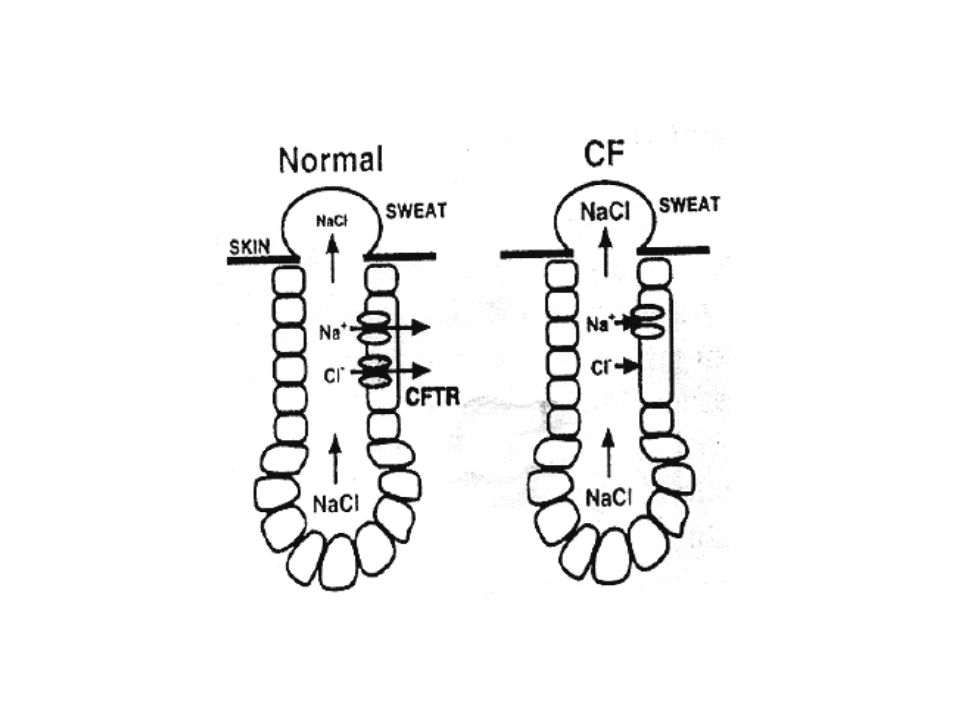
La proteina: CFTR Cystic Fibrosis Transmembrane Conductance Regulator è il nome della proteina che ha la funzione di “canale dei cloruri” cioè permette il passaggio del cloro attraverso la membrana cellulare a spese di ATP. La mutazione del gene che produce questa proteina determina il mancato trasporto di cloro e acqua da parte delle cellule secernenti.

10
Distribuzione e funzioni di CFTR
CFTR espresso in: polmone (bassa densità) rene (alta densità) ghiandole salivari ghiandole sudoripare intestino cuore testicoli In cellule normali: CFTR forma una conduttanza per il Cl- Determina la perdita di Cl- e la secrezione di fluidi Determina il riassorbimento di Cl- dai dotti salivari e sudoripari.
 rene (alta densità) ghiandole salivari. ghiandole sudoripare. intestino. cuore. testicoli. In cellule normali: CFTR forma una conduttanza. per il Cl- Determina la perdita di Cl- e la secrezione di fluidi. Determina il riassorbimento di Cl- dai dotti salivari e sudoripari.")
11
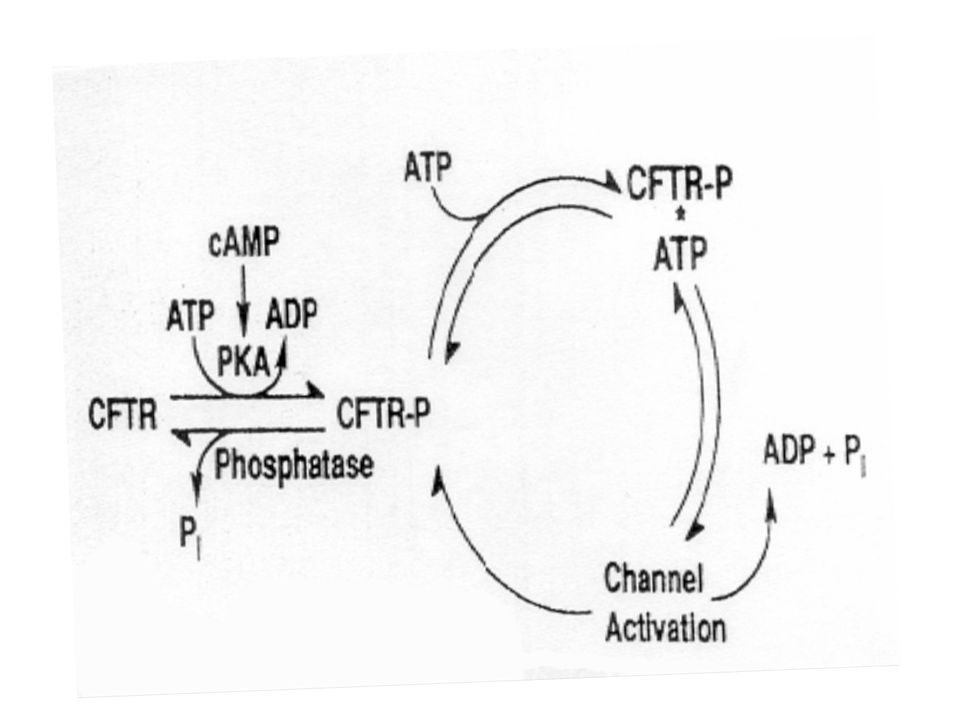
Regolazione di CFTR CFTR si apre se:
Mutazioni nel dominio R possono alterare la regolazione chinasi-dipendente lasciando il canale aperto oppure chiuso N C R NBD1 NBD2 CFTR si apre se: È fosforilato nel dominio R da PKA, PKC, PKG Si lega ATP (con e senza idrolisi) a NBD1 & NBD2
 a NBD1 & NBD2.")
13
CFTR e Secrezione Cotrasportatore Na/K/2Cl aumenta [Cl]i
LUME cAMP Cl- + H2O Canali del K+ basolaterali mantengono il pot. di membrana negativo Na+ CFTR (apicale) pemette la perdita di Cl- Na+ K+ Alta [Cl-] Na+ segue paracellularmente Na+ K+ 2 Cl- Acqua abbandona per osmosi SANGUE
![CFTR e Secrezione Cotrasportatore Na/K/2Cl aumenta [Cl]i](http://slideplayer.it/slide/6117963/19/images/13/CFTR+e+Secrezione+Cotrasportatore+Na%2FK%2F2Cl+aumenta+%5BCl%5Di.jpg "LUME. cAMP. Cl- + H2O. Canali del K+ basolaterali mantengono il pot. di membrana negativo. Na+ CFTR (apicale) pemette la perdita di Cl- Na+ K+ Alta [Cl-] Na+ segue paracellularmente. Na+ K+ 2 Cl- Acqua abbandona per osmosi. SANGUE.")
14
CFTR e Riassorbimento Cl- (A) Se la [Cl-]LUME è elevata
Sodio - Il gradiente di Na+ ne favorisce il suo ingresso (amiloride-sensibile) -30 mV Vt Na+ K+ Lume sangue - + La perdita di Na+ lascia il lume negativo (rispetto al sangue) CFTR Cl- PKA + H2O Il potenziale transepiteliale (Vt) fornisce il gradiente elettrochimico per l’assorbimento di Cl- Na+ Canale del Na amiloride-sensibile -40 mV [Cl]Lume >50 mM
![CFTR e Riassorbimento Cl- (A) Se la [Cl-]LUME è elevata](http://slideplayer.it/slide/6117963/19/images/14/CFTR+e+Riassorbimento+Cl-+%28A%29+Se+la+%5BCl-%5DLUME+%C3%A8+elevata.jpg "Sodio - Il gradiente di Na+ ne favorisce il suo ingresso (amiloride-sensibile) -30 mV. Vt. Na+ K+ Lume. sangue. - + La perdita di Na+ lascia il lume negativo (rispetto al sangue) CFTR. Cl- PKA. + H2O. Il potenziale transepiteliale (Vt) fornisce il gradiente elettrochimico per l’assorbimento di Cl- Na+ Canale del Na amiloride-sensibile. -40 mV. [Cl]Lume. >50 mM.")
15
CFTR e Riassorbimento Vt X (B) Se la [Cl-]LUME è bassa
<50 mM (B) Se la [Cl-]LUME è bassa -40 mV Vt Si ha ingresso di Na+ lume ancora negativo (Vt) Na+ K+ Lumen Blood CFTR defosforilato X Ma Vt non fornisce più il gradiente elettrochimico per l’assorbimento di Cl- Il risultante gradiente di HCO3- è usato per assorbire il Cl- Cl- HCO3- Lo scambiatore Na+/H+ acidifica il lume Na+ H+ Canale del Na+ amiloride-sensibile Na+
![CFTR e Riassorbimento Vt X (B) Se la [Cl-]LUME è bassa](http://slideplayer.it/slide/6117963/19/images/15/CFTR+e+Riassorbimento+Vt+X+%28B%29+Se+la+%5BCl-%5DLUME+%C3%A8+bassa.jpg "<50 mM. (B) Se la [Cl-]LUME è bassa. -40 mV. Vt. Si ha ingresso di Na+ lume ancora negativo (Vt) Na+ K+ Lumen. Blood. CFTR defosforilato. X. Ma Vt non fornisce più il gradiente elettrochimico per l’assorbimento di Cl- Il risultante gradiente di HCO3- è usato per assorbire il Cl- Cl- HCO3- Lo scambiatore Na+/H+ acidifica il lume. Na+ H+ Canale del Na+ amiloride-sensibile. Na+")
17
CFTR-DF508 normalmente non raggiunge la membrana plasmatica;
WT CFTR CFTR-DF508

18
Patogenesi Accumulo di secrezioni mucose vischiose a carico delle ghiandole, in particolare: Pancreas Ghiandole mucipare di polmoni e intestino

19
FIBROSI CISTICA – Fenotipo
1. Il muco secreto nei polmoni intrappola polvere e batteri; le cellule ciliate muovono il muco verso la bocca. Nella FC, il muco è denso, non viene mosso, resta nei polmoni provocando tosse persistente e ricorrenti infezioni polmonari 2. Il pancreas secerne enzimi digestivi nell’intestino Nella FC, esso si ostruisce provocando cisti e assumendo un aspetto fibroso – da cui il nome: fibrosi cistica del pancreas 3. Intestino non riceve abbastanza enzimi digestivi specifici per i grassi provocando denutrizione ed eccesso di grassi nelle feci (steatorrea). 4. I dotti deferenti nel maschio si ostruiscono causando sterilità. 5. Ghiandole sudoripare: secernono sali per poter indurre un flusso d’acqua sulla superficie della pelle per raffreddare; molta parte di questo sale è ripreso dall’organismo. Nella FC, il sale non viene ripreso depositandosi sulla pelle e riducendosi nell’organismo.
. 4. I dotti deferenti nel maschio si ostruiscono causando sterilità. 5. Ghiandole sudoripare: secernono sali per poter indurre un flusso d’acqua sulla superficie della pelle per raffreddare; molta parte di questo sale è ripreso dall’organismo. Nella FC, il sale non viene ripreso depositandosi sulla pelle e riducendosi nell’organismo.")
20
Sintomatologia Malattia polmonare cronica Insufficienza pancreatica
Perdita aumentata di elettroliti nel sudore (alla nascita una complicanza frequente è l’ileo da meconio)
")
21
La Fibrosi Cistica si manifesta con forme cliniche molto differenziate:
Polisintomatiche, gravi (tipiche) Oligosintomatiche Monosintomatiche, lievi (atipiche)
 Oligosintomatiche. Monosintomatiche, lievi (atipiche)")
22
Terapie quotidiane Aerosol, fisioterapia, attività fisica e periodiche terapie antibiotiche (per limitare i danni a livello polmonare) Enzimi pancreatici e vitamine ad ogni pasto (per facilitare il processo digestivo e assimilativo) Tale regime terapeutico coinvolge almeno un familiare per lunghi anni, finché il paziente non è in grado di assumere la completa responsabilitàdelle cure.
 Tale regime terapeutico coinvolge almeno un familiare per lunghi anni, finché il paziente non è in grado di assumere la completa responsabilitàdelle cure.")
23
Oggi le aumentate conoscenze sulla Fibrosi Cistica, lo screening neonatale e soprattutto l’aumentata efficacia delle terapie mediche, fanno sì che i bambini con la Fibrosi Cistica, abbiano uno sviluppo somatico “normale”e che gli Adulti affetti da Fibrosi Cistica abbiano un buono stato clinico. Il miglioramento dell’intervento medico ha allungato la vita di questi pazienti e sicuramente lo farà ancora di più in futuro.

24
Le nuove terapie per i polmoni
Gene mutato Terapia genica Farmaci “correttori” “potenziatori” Proteina CFTR difettosa Anomalo trasporto cloro e sodio Modulatori dei canali Infezione Nuovi antibiotici ed antiinfiammatori Infiammazione Danno

25
Terapia genica: i vettori
Gene CFTR Virus Adeno-associati Adenovirus Liposomi

26
Terapia genica: gli ostacoli
Instaurarsi di processi infiammatori (tossicità) Aumento di anticorpi neutralizzanti (ripetizione delle dosi?) muco = barriera Penetrazione nella cellula (quali recettori?) Accesso al nucleo e resintesi di proteina CFTR funzionante
 Aumento di anticorpi neutralizzanti. (ripetizione delle dosi ) muco = barriera. Penetrazione nella cellula (quali recettori ) Accesso al nucleo e resintesi di proteina. CFTR funzionante.")
27
Cellule staminali: terapia cellulare

Presentazioni simili