Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
ADME FARMACOCINETICA ASSORBIMENTO DISTRIBUZIONE
Studio dell’evoluzione temporale delle concentrazioni di un farmaco e dei suoi metaboliti nei diversi fluidi e nei tessuti dell’organismo mediante l’analisi dei processi che regolano: ASSORBIMENTO DISTRIBUZIONE METABOLISMO ELIMINAZIONE ADME

2
OBIETTIVI: Sviluppare nuovi farmaci
Selezionare la via di somministrazione Scegliere la migliore forma farmaceutica Conoscere la capacità di accesso ad organi e tessuti Conoscere le vie metaboliche Caratterizzare i processi di eliminazione Stabilire le relazioni con la risposta farmacologica Migliorare i risultati dei trattamenti

3
CICLO DI UN FARMACO DOPO LA SOMMINISTRAZIONE
DOSE ORALE DOSE I.V. TRATTO G.I. CIRCOLAZIONE SISTEMICA DISTRIBUZIONE PERIFERICA FEGATO ORGANO BERSAGLIO CLEARANCE RECETTORE EFFETTO FARMACOLOGICO

4
Proteine e, in generale, grosse molecole
Passaggio dei farmaci attraverso le membrane biologiche in funzione delle loro caratteristiche chimico-fisiche Caratteristiche del farmaco Passaggio attraverso le membrane biologiche PROCESSO PASSIVO Sostanze idrosolubili, non ionizzabili, con diametro molecolare inferiore a 4 Å (acqua, urea, alcool) - Filtrazione attraverso i pori Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata. Il trasferimento dipende dal pKa della sostanza e dal gradiente di pH ai due lati della membrana MECCANISMO DI TRASPORTO Sostanze idrosolubili non ionizzate con diametro superiore a 4 Å (glucosio) - Diffusione facilitata senza dispendio energetico per mezzo di un trasportatore Acidi e basi organiche ionizzate -Trasporto attivo con dispendio energetico mediante un trasportatore Proteine e, in generale, grosse molecole - Fagocitosi e pinocitosi (trasporto vescicolare)
 - Filtrazione attraverso i pori. Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata. Il trasferimento dipende dal pKa della sostanza e dal gradiente di pH ai due lati della membrana. MECCANISMO DI TRASPORTO. Sostanze idrosolubili non ionizzate con diametro superiore a 4 Å (glucosio) - Diffusione facilitata senza dispendio energetico per mezzo di un trasportatore. Acidi e basi organiche ionizzate. -Trasporto attivo con dispendio energetico mediante un trasportatore. Proteine e, in generale, grosse molecole. - Fagocitosi e pinocitosi (trasporto vescicolare)")
5
DELLA FORMA NON-IONIZZATA
LA MAGGIOR PARTE DEI FARMACI E’ ASSORBITA PER DIFFUSIONE PASSIVA DELLA FORMA NON-IONIZZATA

6
COEFFICIENTE DI RIPARTIZIONE
E’ molto importante la solubilità del farmaco nel doppio strato lipidico, misurata dal coefficiente di ripartizione che indica come un farmaco si distribuisce in una soluzione contenente H2O e olio: Se > il farmaco è lipofilo e diffonde facilmente Se < il farmaco è idrofilo e non diffonde facilmente COEFFICIENTE DI [farmaco] nella fase oleosa = RIPARTIZIONE [farmaco] nella fase acquosa Il coefficiente di ripartizione non è un parametro fisso, ma può variare in diverse situazioni, per esempio: per metabolismo del farmaco la maggior parte dei farmaci sono acidi o basi deboli, quindi il coefficiente varia a seconda del pH dell’ambiente nel quale si trovano (questa variabile può essere sfruttata anche per aumentare la velocità di eliminazione: alcalinizzazione delle urine in caso di avvelenamento da barbiturici)
")
7
Un importante fattore di cui
tener conto in relazione alla permeazione delle membrane è che molti farmaci sono acidi o basi deboli e, dunque, esistono sia in forma ionizzata che non ionizzata.

8
PRINCIPALI VIE DI SOMMINISTRAZIONE ED ELIMINAZIONE
DEL FARMACO

9
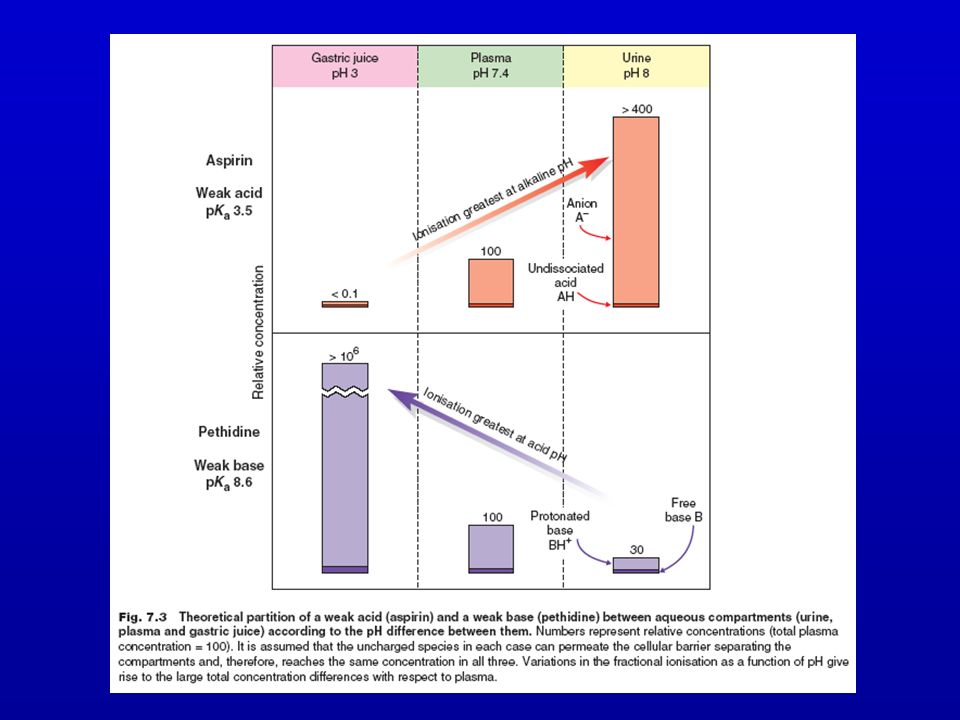
ASSORBIMENTO GASTRICO DI UN SOSTANZA ACIDA
(es.: acido acetilsalicilico pKa = 3,4) Stomaco: pH = 1,4 Plasma: pH = 7,4
 Stomaco: pH = 1,4. Plasma: pH = 7,4.")
11
Riassumendo, l’entità dell’assorbimento di un farmaco dipende dal suo pKa, dalla sua lipofilia e dal pH del mezzo. Questi 3 parametri sono tra loro correlati nella cosiddetta ipotesi della ripartizione in funzione del pH Il tratto GI, al pari di altre membrane, si comporta come una barriera lipofila Acidi e basi sono assorbiti di preferenza in forma indissociata La maggior parte dei farmaci è assorbita per diffusione passiva La velocità di assorbimento e la quantità di farmaco assorbita sono correlate al coefficiente di ripartizione: maggiore è la liposolubilità maggiore è l’assorbimento Acidi deboli e farmaci neutri possono essere assorbiti nello stomaco, ma non le basi.

12
FATTORI CRUCIALI PER L’ASSORBIMENTO
DEI FARMACI Caratteristiche del farmaco: massa molecolare, stato fisico, carica, stabilità, solubilità…. Proprietà dell’organismo: morfologia e dimensioni della superficie assorbente, perfusione dell’area assorbente, specie, razza, età, stato nutrizionale, stato di salute……… Caratteristiche dell’esposizione: dose, via di somministrazione, durata del contatto con la superficie assorbente…. Fattori esogeni: formulazione, interazione con altre sostanze, condizioni fisiche (es. temperatura)…..
…..")
13
La velocità di assorbimento varia a seconda della via di somministrazione utilizzata
La concentrazione plasmatica di un farmaco nell’unità di tempo dipende dalla differenza tra la quantità assorbita e la quantità eliminata Il picco di concentrazione plasmatica di un farmaco dipende dalla velocità di assorbimento: più lento è l’assorbimento, più basso è il picco plasmatico

14
FATTORI CHE CONDIZIONANO L’ASSORBIMENTO
GASTROINTESTINALE Legge di azione di massa Equazione di Henderson-Hasselbach Fase farmaceutica (disintegrazione e dissoluzione) Area superficiale di assorbimento Velocità del flusso ematico Resistenza al pH gastrico, agli enzimi dello stomaco, dell’intestino e della flora intestinale Trasporto specializzato Circolo enteroepatico
 Area superficiale di assorbimento. Velocità del flusso ematico. Resistenza al pH gastrico, agli enzimi dello stomaco, dell’intestino. e della flora intestinale. Trasporto specializzato. Circolo enteroepatico.")
15
L’assorbimento di farmaci dall’intestino è una funzione del pKa, per acidi e basi

16
VARIABILITA’ FARMACOCINETICA
Il grafico mostra l’andamento della concentrazione plasmatica di digossina in seguito a somministrazione allo stesso soggetto di 4 formulazioni commerciali di digossina prodotta da 3 ditte diverse (B e C sono formulazioni prodotte dalla stessa ditta) 3 2 1 concentrazione plasmatica digossina (ug/ml) A B C D tempo (ore) LA DIVERSA BIODISPONIBILITA’ PROVOCA PICCHI PLASMATICI DIVERSI SIA IN TERMINI QUANTITATIVI CHE TEMPORALI
 concentrazione plasmatica digossina (ug/ml) A. B. C. D tempo (ore) LA DIVERSA BIODISPONIBILITA’ PROVOCA PICCHI PLASMATICI DIVERSI SIA IN TERMINI QUANTITATIVI CHE TEMPORALI.")
17
Riassumendo…….. Farmaci a bassa solubilità lipidica, inclusi acidi e basi forti, sono generalmente poco assorbiti dall’intestino. Alcuni farmaci sono assorbiti grazie a sistemi di trasporto (carrier protein). L’assorbimento intestinale dipende da molti fattori: - motilità gastrointestinale; - pH gastrointestinale; - dimensione delle particelle; - interazioni fisico-chimiche con i contenuti intestinali (ad es. l’interazione tra Ca2+ e tetracicline).
. L’assorbimento intestinale dipende da molti fattori: - motilità gastrointestinale; - pH gastrointestinale; - dimensione delle particelle; - interazioni fisico-chimiche con i contenuti intestinali. (ad es. l’interazione tra Ca2+ e tetracicline).")
18
Riassumendo…….. La biodisponibilità è la frazione di dose ingerita che accede alla circolazione sistemica. Può essere bassa per assorbimento incompleto, o perché il farmaco viene metabolizzato nell’intestino o nel fegato prima di raggiungere la circolazione sistemica. La bioequivalenza implica che, se una formulazione di un farmaco viene sostituita con un’altra, non si dovrà riscontrare alcun effetto indesiderato e/o imprevisto.

19
LA BARRIERA EMATOENCEFALICA
L’endotelio dei vasi cerebrali ha caratteristiche morfologiche e funzionali tali da costituire la barriera ematoencefalica che impedisce l’ingresso nel liquido interstiziale cerebrale di qualunque sostanza incapace di diffondere liberamente attraverso le membrane

20
La Barriera Emato-Encefalica Contribuisce all’omeostasi del SCN
I CAPILLARI DEL SNC SONO SIGILLATI DA GIUNZIONI SERRATE I gas respiratori ed alcune molecole liposolubili diffondono liberamente. Le sostanze nutritive vengono trasportate attivamente. Quelle che potrebbero turbare l’omeostasi del SNC vengono bloccate. GLI ASTROCITI SVOLGONO UN RUOLO FONDAMENTALE NEL PROMUOVERE LE GIUNZIONI SERRATE

21
Nel SNC possono quindi penetrare soltanto:
farmaci con un adeguato coefficiente di distribuzione (direttamente dipendente dal coefficiente di ripartizione) farmaci capaci di utilizzare i sistemi di trasporto presenti a livello della barriera ematoencefalica Lo stato di impermeabilità è ridotto a livello dei plessi coroidei e di altre regioni periventricolari, dove hanno normalmente luogo i processi di filtrazione e secrezione. Inoltre, l’impermeabilità della barriera è ridotta in corso di infiammazione e infezione (meningite).
 farmaci capaci di utilizzare i sistemi di trasporto presenti a livello della barriera ematoencefalica. Lo stato di impermeabilità è ridotto a livello dei plessi coroidei e di altre regioni periventricolari, dove hanno normalmente luogo i processi di filtrazione e secrezione. Inoltre, l’impermeabilità della barriera è ridotta in corso di infiammazione e infezione (meningite).")
22
ASSORBIMENTO POLMONARE (gas, vapori e liquidi volatili)
L’inalazione è la via di somministrazione per anestetici gassosi e sostanze volatili. Il polmone agisce come sito di somministrazione e di eliminazione I farmaci usati per i loro effetti sul polmone sono dati per via inalatoria, di solito come aerosol Glucocorticoidi (beclometasone) e broncodilatatori (salbutamolo) raggiungono elevate concentrazioni locali con minimizzazione degli effetti collaterali. Cellule epiteliali degli alveoli ed endotelio dei capillari sono ampiamente fenestrati Flusso ematico elevato Coefficiente di ripartizione liquido/gas
 e broncodilatatori (salbutamolo) raggiungono elevate concentrazioni locali con minimizzazione degli. effetti collaterali. Cellule epiteliali degli alveoli ed endotelio dei capillari. sono ampiamente fenestrati. Flusso ematico elevato. Coefficiente di ripartizione liquido/gas.")
23
DISTRIBUZIONE DEI FARMACI
Processo mediante il quale un farmaco passa da un distretto corporeo all’altro fino a raggiungere il sito d’azione

24
DISTRIBUZIONE Processo di ripartizione in tre fasi liquide: Plasma
Fluidi extracellulari Fluidi intracellulari Il plasma rappresenta circa il 4.5% del peso corporeo. I fluidi extracellulari comprendono: fluido interstiziale (16%) e linfa (1.2%). I fluidi intracellulari (30-40%) sono la somma di tutti i contenuti fluidi delle cellule. I fluidi transcellulari (2.5%) comprendono i fluidi cerebrospinale, intraoculare, peritoneale, pleurale e sinoviale.
 e linfa (1.2%). I fluidi intracellulari (30-40%) sono la somma di tutti i contenuti. fluidi delle cellule. I fluidi transcellulari (2.5%) comprendono i fluidi cerebrospinale, intraoculare, peritoneale, pleurale e sinoviale.")
25
Le molecole di farmaco esistono in forma legata o libera in ciascun
compartimento, ma solo le molecole libere possono muoversi tra compartimenti L’equilibrio di distribuzione tra i compartimenti dipende da: - permeabilità tra le barriere tissutali; - legame con i compartimenti; - ripartizione dovuta al pH; - ripartizione grasso:acqua

26
Fattori che influenzano la distribuzione
di un farmaco Caratteristiche fisico-chimiche del farmaco Legame della molecola alle proteine plasmatiche (protein binding) Irrorazione degli organi Affinità specifica dei tessuti
 Irrorazione degli organi. Affinità specifica dei tessuti.")
27
Alla somministrazione All’equilibrio
Farmaco idrosolubile Plasma Cellule Farmaco liposolubile Plasma Cellule

28
Legame alle proteine (protein binding) Soprattutto alle albumine
Il farmaco legato non attraversa le membrane Equilibrio dinamico tra parte libera e legata

29
L’albumina plasmatica è la più importante; anche b-globulina e a-glicoproteina legano alcuni farmaci. L’albumina plasmatica lega soprattutto farmaci acidi (2:1). I farmaci basici vengono legati dalla b-globulina e dalla a-glicoproteina. Un protein binding estensivo rallenta l’eliminazione del farmaco. La competizione tra farmaci per il protein binding può causare interazioni clinicamente rilevanti.
. I farmaci basici vengono legati dalla b-globulina e dalla a-glicoproteina. Un protein binding estensivo rallenta l’eliminazione del farmaco. La competizione tra farmaci per il protein binding può causare interazioni clinicamente rilevanti.")
30
Albumina -------------------- Acidi a1-glicoproteina acida ----- Basi
Plasma Acqua Extracellulare Proteine Plasmatiche Proteine Tissutali Drug Albumina Acidi a1-glicoproteina acida Basi Globuline Steroidi

31
Farmaci molto legati... …..alle albumine o alle a-glicoproteine: FANS
warfarin (anticoagulante) furosemide (diuretico) chinidina (antiaritmico) diazepam (antiepilettico) Propranololo (antagonista b-adrenergico)
 furosemide (diuretico) chinidina (antiaritmico) diazepam (antiepilettico) Propranololo (antagonista b-adrenergico)")
32
Fattori che modificano il legame farmaco-proteico
Modifica del tasso di proteine plasmatiche dovuta a: Insufficienza epatica Insufficienza renale Enteropatie Parassitosi Ustioni Se aumenta la quota libera: Aumento dell’effetto farmacologico Aumento della velocità di eliminazione

33
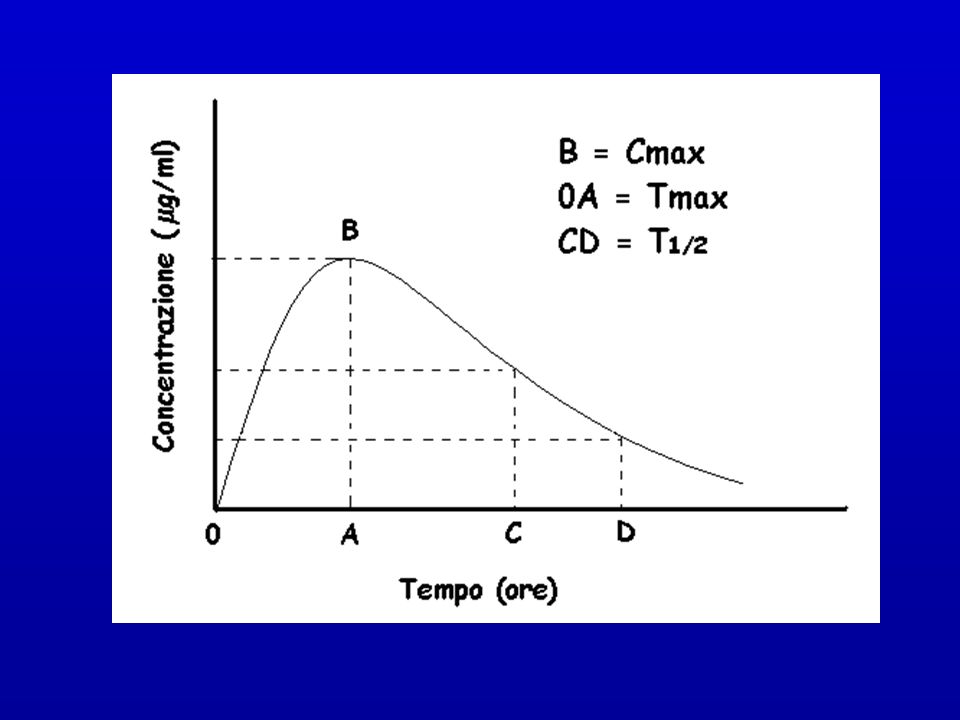
PRINCIPALI PARAMETRI FARMACOCINETICI
Cmax: concentrazione massima Tmax: tempo per raggiungere la Cmax AUC: area sotto la curva F%: biodisponibilità t½: tempo necessario perché la concentrazione plasmatica si riduca della metà Vd: volume di distribuzione Cl: clearance (quantità di farmaco eliminata nell’unità di tempo)
")
35
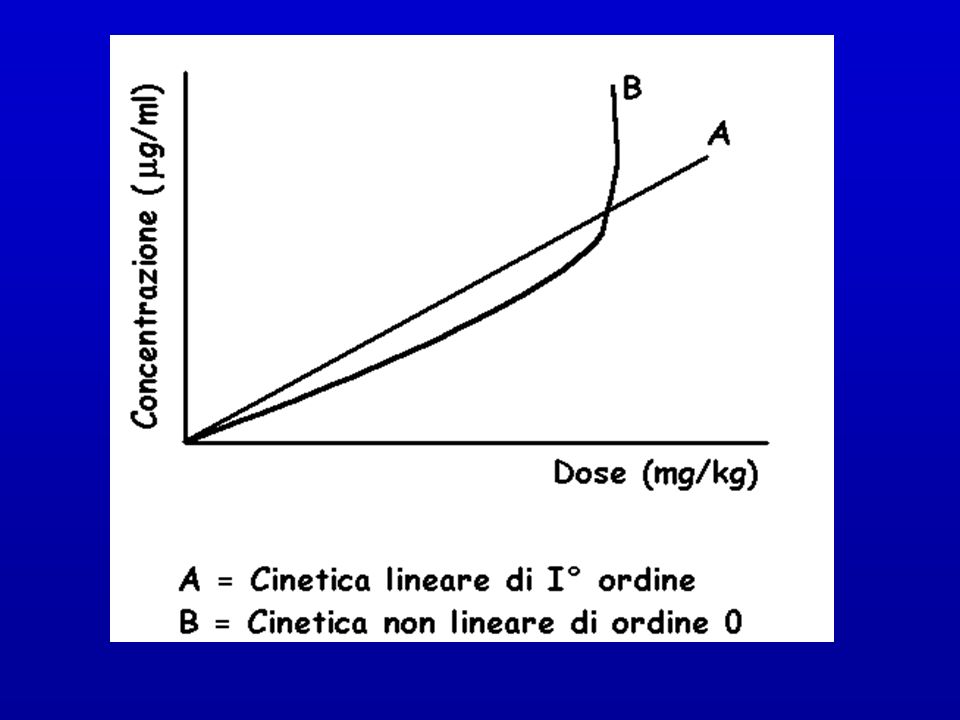
CINETICA DI ORDINE ZERO
CINETICA DI I° ORDINE La variazione di tutti i processi connessi con l’impiego di un farmaco è direttamente proporzionale alla concentrazione di farmaco nel sistema (plasma) CINETICA DI ORDINE ZERO Al di sopra di un certo valore di concentrazione l’assorbimento, la biotrasformazione e l’eliminazione di un farmaco non risultano più proporzionali alla sua concentrazione nel plasma Questo fenomeno può essere dovuto al raggiungimento di condizioni di saturazione dei meccanismi enzimatici e di eliminazione, per cui si può avere un improvviso e marcato aumento della concentrazione nel plasma per piccoli aumenti del dosaggio
 CINETICA DI ORDINE ZERO. Al di sopra di un certo valore di concentrazione l’assorbimento, la biotrasformazione e l’eliminazione di un farmaco non risultano. più proporzionali alla sua concentrazione nel plasma. Questo fenomeno può essere dovuto al raggiungimento di condizioni. di saturazione dei meccanismi enzimatici e di eliminazione, per cui. si può avere un improvviso e marcato aumento della concentrazione. nel plasma per piccoli aumenti del dosaggio.")
37
Dose Singola Orale In seguito a somministrazione extravascolare (via orale) di un farmaco, la sua concentrazione plasmatica crescerà fino a un valore massimo (Cmax) e poi inizierà a diminuire. Man mano che aumenta la concentrazione plasmatica, inizia anche il processo di eliminazione. Il tempo che impiega un farmaco somministrato in singola dose orale per raggiungere la Cmax è detto tmax
 e poi inizierà a diminuire. Man mano che aumenta la concentrazione plasmatica, inizia anche. il processo di eliminazione. Il tempo che impiega un farmaco somministrato in singola dose. orale per raggiungere la Cmax è detto tmax.")
38
Somministrazione ripetuta
E’ usata per mantenere la concentrazione plasmatica di un farmaco nella finestra terapeutica. La conc. raggiunge un plateau, valore al quale la velocità di assorbimento è uguale a quella di eliminazione. I rettangoli bianchi indicano la quantità di farmaco presente nell’organismo nel momento immediatamente precedente la somministrazione della dose; i rettangoli gialli rappresentano la dose e la somma indica la quantità di farmaco presente nell’organismo nel momento immediatamente successivo ad ogni somministrazione.

39
Biodisponibilità La fisiologia dell’assorbimento del farmaco dal tratto GI ha effetto diretto sulla BIODISPONIBILITA’ del farmaco. La biodisponibilità è definita come la frazione di dose di un farmaco (F) che entra nella circolazione sistemica: F = Quantità di farmaco nella circolazione sistemica/ Dose somministrata Poiché l’area sotto la curva “concentrazione plasmatica vs. tempo” (AUC) è una misura della concentrazione del farmaco nella circolazione sistemica, la biodisponibilità è di solito definita come: F = AUC/Dose
 che entra nella circolazione sistemica: F = Quantità di farmaco nella circolazione sistemica/ Dose somministrata. Poiché l’area sotto la curva concentrazione plasmatica vs. tempo (AUC) è una misura della concentrazione del farmaco nella. circolazione sistemica, la biodisponibilità è di solito definita come: F = AUC/Dose.")
41
Riassumendo: la BIODISPONIBILITA’ di un farmaco rappresenta: 1) la frazione della dose somministrata che raggiunge la circolazione sistemica 2) la velocità con cui questo avviene. Somministrazione i.v.: la biodisponibilità è, per definizione, 100%. Altre vie di somministrazione: 0% <biodisponibilità <100%.
 la velocità con cui questo avviene. Somministrazione i.v.: la biodisponibilità è, per definizione, 100%. Altre vie di somministrazione: 0% <biodisponibilità <100%.")
42
Tempo di emivita (t1/2) Necessario per determinare:
Intervallo tra dosi successive di farmaco Durata dell’effetto benefico o tossico Tempi di sospensione Per le reazioni di I ordine t1/2 SEMPRE costante t1/2 = / K

43
Emivita di un farmaco N° di t½ Frazione di farmaco rimanente 1 2 3 4 5
1 2 3 4 5 6 7 8 9 10 100% 50% 25% 12.5% 6.25% 3.125% 1.56% 0.78% 0.39% 0.195% 0.0975% *** Sono necessarie 10 emivite per eliminare il 99,9%***

44
Volume di distribuzione
DEFINIZIONE: E’ una costante di proporzionalità che correla la quantità di farmaco presente in un certo momento nell’organismo alla sua concentrazione plasmatica nello stesso istante. Questo valore, frutto di un calcolo, non corrisponde direttamente ad una parte anatomica o fisiologica dell’organismo e può essere molto più grande del volume dell’acqua corporea complessiva. Perciò è detto volume “apparente” di distribuzione.

45
Volume di distribuzione
Volume di fluido richiesto per contenere la quantità totale, Q, di farmaco nel corpo alla stessa concentrazione presente nel plasma, Cp Vd = Q/Cp

46
Volume di distribuzione
volume “apparente” nel quale è sciolto il farmaco Indica la distribuzione del farmaco nell’organismo È influenzato dalla capacità di attraversare le membrane biologiche Grado di ionizzazione Liposolubilità Peso molecolare

47
Volume di distribuzione
Vd = Q/Cp Più la concentrazione plasmatica di un farmaco è elevata rispetto alla dose iniziale, più il valore numerico del Vd sarà piccolo, ad indicare che il farmaco è scarsamente Distribuito- Al contrario, una bassa concentrazione plasmatica rispetto alla dose indicherà che il farmaco si è distribuito in altri distretti dell’organismo ed avrà quindi un elevato volume di distribuzione.

48
Volume di distribuzione
VOLUME DI DISTRIBUZIONE DI ALCUNI FARMACI DRUG Vd (mL/kg) cocaina 140 amoxicillina 700 amitriptilina amiodarone ~5000 Intracellulare = 370 mL/kg Volume di Acqua Totale = 670 mL/kg Extracellulare = 300 mL/kg Volume Plasmatico = 40 mL/kg
 cocaina 140. amoxicillina 700. amitriptilina amiodarone ~5000. Intracellulare = 370 mL/kg. Volume di Acqua Totale = 670 mL/kg. Extracellulare = 300 mL/kg. Volume Plasmatico = 40 mL/kg.")
49
MODELLI FARMACOCINETICI
L’accuratezza dei risultati di uno studio farmacocinetico richiede l’uso di metodi matematici. Per poter applicare tali metodi al comportamento di un farmaco in un sistema biologico complesso, è necessario usare sistemi “modello”. Il “modello” simula le relazioni tra assorbimento, distribuzione, risposta e eliminazione del farmaco in varie sezioni del sistema biologico. Il modello permette al chimico farmaceutico di usare equazioni matematiche per descrivere le relazioni tra concentrazioni di un farmaco in vari tessuti e, come risultato, predire la concentrazione in un tessuto in seguito a ben definite modalità di somministrazione. VANTAGGI: correlazioni tra dosi e risposte farmacologiche e/o tossiche SVANTAGGI: eccessiva semplificazione rispetto alla complessità dei sistemi biologici

50
COMPARTIMENTO: tessuto o insieme di tessuti diversi che hanno
un comportamento analogo nei confronti di una determinata sostanza MODELLO MONOCOMPARTIMENTALE: ogni variazione che avviene nei livelli plasmatici di un farmaco si riflette proporzionalmente nei livelli tissutali MODELLO BICOMPARTIMENTALE: la concentrazione del farmaco diminuisce rapidamente dal plasma e dai tessuti più perfusi (compartimento centrale) e si distribuisce più lentamente nei tessuti meno perfusi (compartimento periferico). Si distingue una fase di distribuzione e una fase di eliminazione. MODELLO TRICOMPARTIMENTALE: un terzo compartimento periferico è costituito dai tessuti più profondi scarsamente perfusi. Si distinguono una fase di distribuzione, una di eliminazione rapida ed una di eliminazione lenta.
 e si distribuisce più lentamente nei tessuti. meno perfusi (compartimento periferico). Si distingue una fase di distribuzione e una fase di eliminazione. MODELLO TRICOMPARTIMENTALE: un terzo compartimento. periferico è costituito dai tessuti più profondi scarsamente perfusi. Si distinguono una fase di distribuzione, una di eliminazione rapida. ed una di eliminazione lenta.")
51
MODELLO MONOCOMPARTIMENTALE

52
MODELLO MONOCOMPARTIMENTALE

53
MODELLO BICOMPARTIMENTALE

54
INDIVIDUALIZZATA E RAZIONALE
Scopo finale... UNA TERAPIA INDIVIDUALIZZATA E RAZIONALE

Presentazioni simili