Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Ottimizzazione Lead Ottimizzare le sue caratteristiche farmacologiche Ottimizzazione delle interazione con il target Ottimizzazione dell’accesso al target Farmacodinamica Farmacocinetica Chimica Farmaceutica

2
Ottimizzazione Lead Ottimizzazione dell’accesso al target Modulazione chimica e chimico-fisica I prodotti con la migliore affinità per il bersaglio non è necessariamente il migliore farmaco da usare Spesso prodotti molto attivi in vitro sono inattivi in vivo Caratteristiche farmacocinetiche

3
3 FARMACOCINETICA Le caratteristiche di Assorbimento, Distribuzione, Metabolismo ed Eliminazione/Escrezione (ADME) sono proprietà importanti da considerare nello sviluppo di nuovi agenti terapeutici. Molti composti che pur arrivano ai trials clinici di fase I-III devono essere abbandonati, spesso per problemi riconducibili alle proprietà ADME. Anche la tossicità è un fattore importante ed è a sua volta correlato alle proprietà ADME. Studio dell'attività dei farmaci nel corpo per un periodo di tempo, inclusi i processi di assorbimento, distribuzione, localizzazione nei tessuti, biotrasformazione e escrezione di esso.

4
FARMACODINAMICAFARMACODINAMICA Dose di farmaco somministrato Concentrazione farmaco circolazione sistemica Concentrazione farmaco sito d’azione EFFETTO FARMACOLOGICO Farmaco nel tessuto di distribuzione Farmaco metabolizzato o eliminato RISPOSTA CLINICA Tossicità Efficacia FARMACEUTICAFARMACEUTICA Assorbimento Distribuzione Eliminazione FARMACOCINETICAFARMACOCINETICA

5
5 ASSORBIMENTO DEI FARMACI il processo per mezzo del quale un farmaco passa dal sito di somministrazione alla circolazione sistemica L’assorbimento è un parametro di fondamentale importanza nella progettazione di nuovi farmaci Al momento c’è una netta tendenza ad ottenere candidati farmaci che presentino un buon assorbimento dopo somministrazione orale, nella speranza che ciò si rifletta in una buona biodisponibilità orale

6
Processi che consentono l’assorbimento dei farmaci dell’esposizione: Fattori dipendenti dell’esposizione: dose, via di somministrazione, durata del contatto con la superficie assorbente Fattori dipendenti dal farmaco dimensioni e forma molecolare solubilità in acqua e lipidi grado di ionizzazione solubilità in acqua e lipidi della forma ionizzata Fattori dipendenti dalle membrane cellulari Composizione: % Componenti e natura dei componenti Spessore

7
7 VIE DI SOMMINISTRAZIONE PARENTERALI ENDOVENA 100% assorbimento effetti immediati Utilizzata in emergenza possono essere iniettati grossi volumi si possono somministrare sostanze irritanti diluite (KCl) INTRAMUSCOLO Assorbimento: rapido per le soluzioni acquose lento e prolungato per le preparazioni a lento rilascio Si possono utilizzare volumi moderati si utilizza per somministrare sostanze oleose SOTTOCUTANEA Assorbimento: rapido per le soluzioni acquose lento e prolungato per le preparazioni a lento rilascio è utilizzata per soluzioni insolubili e per l’impianto di pellet solidi
 INTRAMUSCOLO Assorbimento: rapido per le soluzioni acquose lento e prolungato per le preparazioni a lento rilascio Si possono utilizzare volumi moderati si utilizza per somministrare sostanze oleose SOTTOCUTANEA Assorbimento: rapido per le soluzioni acquose lento e prolungato per le preparazioni a lento rilascio è utilizzata per soluzioni insolubili e per l’impianto di pellet solidi")
8
8 VIE DI SOMMINISTRAZIONE ENTERALI PER OS assorbimento variabile, che dipende da molti fattori gli effetti compaiono dopo almeno 45-60 minuti è la via più economica e più sicura possibilità di utilizzo di PREPARAZIONI RETARD RETTALE assorbimento variabile e incompleto ha una latenza d’azione minore rispetto alla via per os SUBLINGUALE assorbimento rapido l’effetto compare dopo pochi minuti utilizzata in emergenza evita l’effetto di primo passaggio

9
Processi di diffusione Le molecole diffonde da una regione ad alta concentrazione ad una regione a più bassa concentrazione MECCANISMI MOLECOLARI ATTRAVERSO CUI PUO’ AVVENIRE IL PASSAGGIO DI FARMACI ATTRAVERSO LA MEMBRANA PLASMATICA

10
CHIMICO-FISICI DELLA MOLECOLA Peso molecolare (pm), Diffusibilitá Liposolubilitá Grado di dissociazione PARAMETRI DIFFUSIONE PASSIVA CARATTERISTICHE FISIOLOGICHE, ANATOMICHE E PATOLOGICHE DEL PAZIENTE
, Diffusibilitá Liposolubilitá Grado di dissociazione PARAMETRI DIFFUSIONE PASSIVA CARATTERISTICHE FISIOLOGICHE, ANATOMICHE E PATOLOGICHE DEL PAZIENTE")
11
DIFFUSIBILITÁ attraverso le membrane è un termine che a sua volta include altri parametri e il tutto viene spiegato con la LEGGE DI FICK. dQ/dT = D x P x A (C1-C2) / e dQ/dT= velocità di diffusione (quantità di farmaco assorbito nell'unità di tempo) D = coefficiente di diffusione (capacità del farmaco di diffondere in base alle sua proprietà) K = coefficiente di ripartizione (cioè se il farmaco è più idrosolubile o liposolubile) A = indica la superficie della zona di assorbimento (C1-C2) = indica la concentrazione del farmaco ai lati della membrane e = indica lo spessore della membrana
 / e dQ/dT= velocità di diffusione (quantità di farmaco assorbito nell unità di tempo) D = coefficiente di diffusione (capacità del farmaco di diffondere in base alle sua proprietà) K = coefficiente di ripartizione (cioè se il farmaco è più idrosolubile o liposolubile) A = indica la superficie della zona di assorbimento (C1-C2) = indica la concentrazione del farmaco ai lati della membrane e = indica lo spessore della membrana.")
12
E’ molto importante IL GRADO DI SOLUBILITÁ del farmaco nel doppio strato lipidico, misurata dal coefficiente di ripartizione che indica come un farmaco si distribuisce in una soluzione contenente H 2 O e olio: Se > 1 il farmaco è lipofilo e diffonde facilmente Se < 1 il farmaco è idrofilo e non diffonde facilmente COEFFICIENTE DI [farmaco] nella fase oleosa = ----------------------------------- RIPARTIZIONE [farmaco] nella fase acquosa COEFFICIENTE DI RIPARTIZIONE
![E’ molto importante IL GRADO DI SOLUBILITÁ del farmaco nel doppio strato lipidico, misurata dal coefficiente di ripartizione che indica come un farmaco si distribuisce in una soluzione contenente H 2 O e olio: Se > 1 il farmaco è lipofilo e diffonde facilmente Se < 1 il farmaco è idrofilo e non diffonde facilmente COEFFICIENTE DI [farmaco] nella fase oleosa = RIPARTIZIONE [farmaco] nella fase acquosa COEFFICIENTE DI RIPARTIZIONE](http://images.slideplayer.it/33/10486658/slides/slide_12.jpg "E’ molto importante IL GRADO DI SOLUBILITÁ del farmaco nel doppio strato lipidico, misurata dal coefficiente di ripartizione che indica come un farmaco si distribuisce in una soluzione contenente H 2 O e olio: Se > 1 il farmaco è lipofilo e diffonde facilmente Se < 1 il farmaco è idrofilo e non diffonde facilmente COEFFICIENTE DI [farmaco] nella fase oleosa = RIPARTIZIONE [farmaco] nella fase acquosa COEFFICIENTE DI RIPARTIZIONE")
13
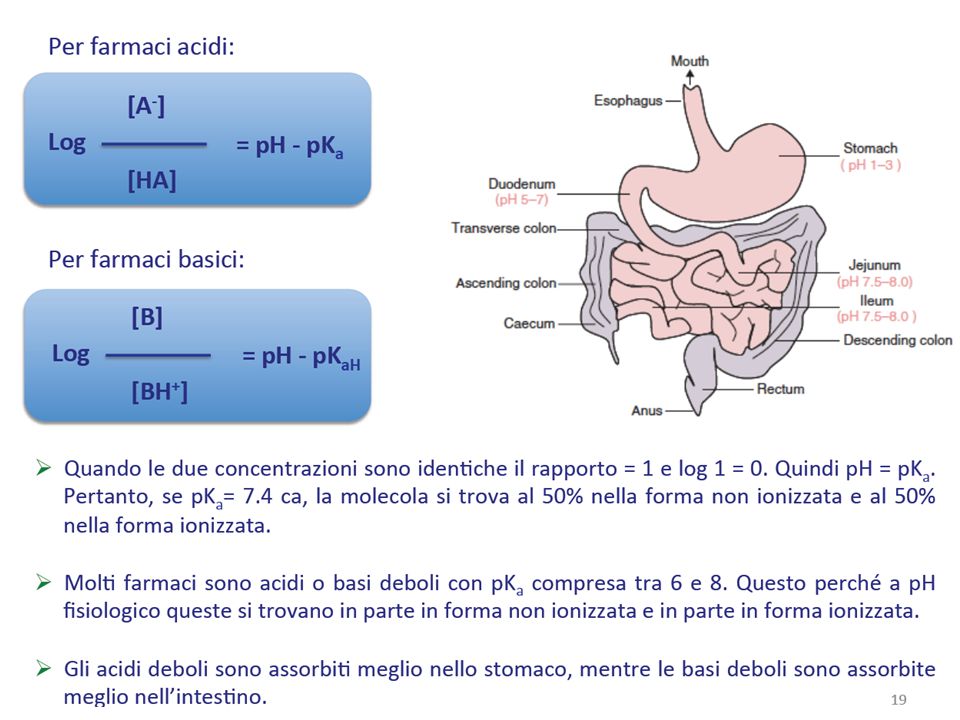
Un importante fattore di cui tener conto in relazione alla permeazione delle membrane è che molti farmaci sono acidi o basi deboli e, dunque, esistono sia in forma ionizzata che non ionizzata. GRADO DI DISSOCIAZIONE

15
Processi di trasporto carrier-mediato Trasporto attivo Il trasporto avviene contro un gradiente di concentrazione e consume ATP Diffusione facilitata Il trasporto avviene secondo un gradiente di concentrazione

16
Esempi di molecole trasportate da carrier ioni inorganici, amminoacidi, monosaccaridi, vitamine, ormoni, penicillamina.

17
L’endocitos i consiste nell’invaginazione di parte della membrana che, dopo aver inglobato la molecola, si richiude formando un vacuolo che migra nella membrana, si riapre dal lato opposto liberando la sostanza. Questo meccanismo avviene per molte macromolecole, vitamine e grassi

19
Riassumendo, l’entità dell’assorbimento di un farmaco dipende dal suo pKa, dalla sua lipofilia e dal pH del mezzo. Questi 3 parametri sono tra loro correlati nella cosiddetta ipotesi della ripartizione in funzione del pH

20
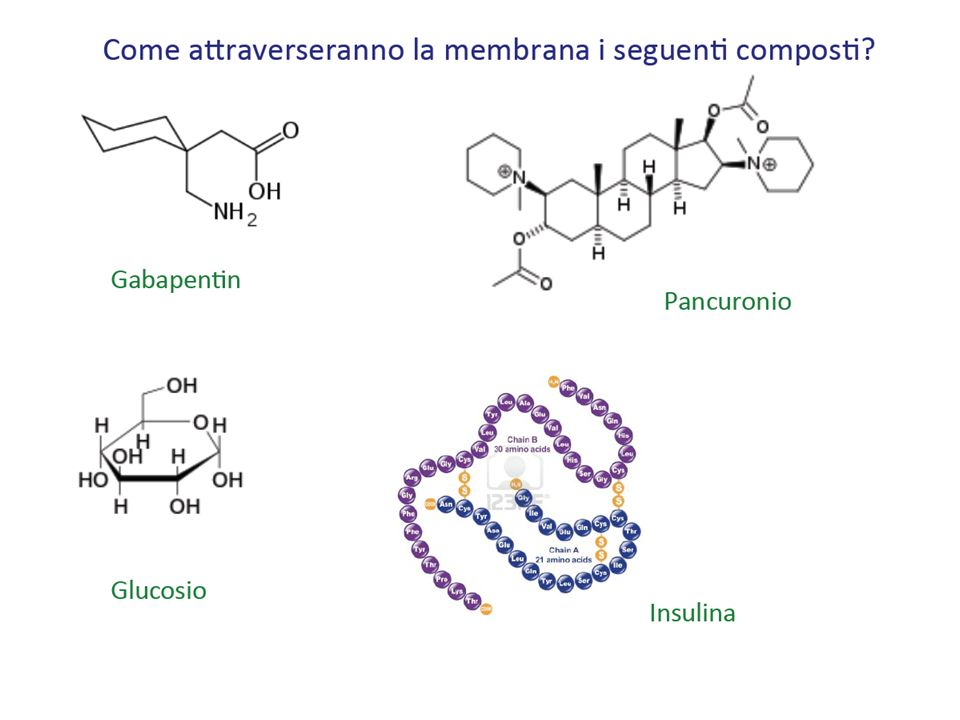
Il tratto gastro intestinaIe, come altre epiteli (orale, rettale, alveolare) si comporta come una barriera lipofila Acidi e basi sono assorbiti di preferenza in forma in dissociata La maggior parte dei farmaci è assorbita per diffusione passiva La velocità di assorbimento e la quantità di farmaco assorbita sono correlate al coefficiente di ripartizione: maggiore è la liposolubilità maggiore è l’assorbimento Acidi deboli e farmaci neutri possono essere assorbiti nello stomaco, ma non le basi. Oltre alle proprietà del farmaco, sono da considerarsi anche le caratteristiche delle membrana o del tessuto da attraversare

21
LA BARRIERA EMATOENCEFALICA La barriera ematoencefalica ha caratteristiche morfologiche e funzionali tali di permette l’ingresso nel liquido interstiziale cerebrale di sostanza capace di diffondere liberamente attraverso le membrane o di essere trasportata attivamente (trasportatori specifici o endocitosi mediata da recettori) Analogamente, vi sono trasportatori deputati alla rimozione di aminoacidi e sostanze tossiche
 Analogamente, vi sono trasportatori deputati alla rimozione di aminoacidi e sostanze tossiche")
22
Nel SNC possono quindi penetrare soltanto: farmaci con un adeguato coefficiente di distribuzione (direttamente dipendente dal coefficiente di ripartizione) farmaci capaci di utilizzare i sistemi di trasporto presenti a livello della barriera ematoencefalica Lo stato di impermeabilità è ridotto a livello dei plessi coroidei e di altre regioni periventricolari, dove hanno normalmente luogo i processi di filtrazione e secrezione. Inoltre, l’impermeabilità della barriera è ridotta in corso di infiammazione e infezione (meningite).
..")
23
ASSORBIMENTO POLMONARE (gas, vapori e liquidi volatili) L’inalazione è la via di somministrazione per anestetici gassosi e sostanze volatili. Il polmone agisce come sito di somministrazione e di eliminazione I farmaci usati per i loro effetti sul polmone sono dati per via inalatoria, di solito come aerosol Glucocorticoidi (beclometasone) e broncodilatatori (salbutamolo) raggiungono elevate concentrazioni locali con minimizzazione degli effetti collaterali. Cellule epiteliali degli alveoli ed endotelio dei capillari sono ampiamente fenestrati Flusso ematico elevato Coefficiente di ripartizione liquido/gas maggiore
 e broncodilatatori (salbutamolo) raggiungono elevate concentrazioni locali con minimizzazione degli effetti collaterali. Cellule epiteliali degli alveoli ed endotelio dei capillari sono ampiamente fenestrati Flusso ematico elevato Coefficiente di ripartizione liquido/gas maggiore.")
24
Può essere attraversata da piccole molecole lipofile Enzimi del metabolismo nel feto poco attivi Escrezione renale non efficiente Tossicità nel feto Placenta

25
DETERMINAZIONE DELL’AUC METODO DEI TRAPEZOIDI PER CALCOLARE L’AUC Area sotto la curva (AUC) misura la quantità di farmaco immodificato che raggiunge la circolazione sistemica dopo somministrazione di una determinata dose, ed è direttamente proporzionale alla quantità di farmaco assorbito PARAMETRI FARMACOCINETICI
 misura la quantità di farmaco immodificato che raggiunge la circolazione sistemica dopo somministrazione di una determinata dose, ed è direttamente proporzionale alla quantità di farmaco assorbito PARAMETRI FARMACOCINETICI")
26
La velocità di assorbimento varia a seconda della via di somministrazione utilizzata La concentrazione plasmatica di un farmaco nell’unità di tempo dipende dalla differenza tra la quantità assorbita e la quantità eliminata Il picco di concentrazione plasmatica di un farmaco dipende dalla velocità di assorbimento: più lento è l’assorbimento, più basso è il picco plasmatico

27
tempo (ore) 1 2 3 4 5 concentrazione plasmatica digossina (ug/ml) A B C D VARIABILITA’ FARMACOCINETICA 32103210 Il grafico mostra l’andamento della concentrazione plasmatica di digossina in seguito a somministrazione allo stesso soggetto di 4 formulazioni commerciali di digossina prodotta da 3 ditte diverse (B e C sono formulazioni prodotte dalla stessa ditta) LA DIVERSA BIODISPONIBILITA’ PROVOCA PICCHI PLASMATICI DIVERSI SIA IN TERMINI QUANTITATIVI CHE TEMPORALI
 concentrazione plasmatica digossina (ug/ml) A B C D VARIABILITA’ FARMACOCINETICA Il grafico mostra l’andamento della concentrazione plasmatica di digossina in seguito a somministrazione allo stesso soggetto di 4 formulazioni commerciali di digossina prodotta da 3 ditte diverse (B e C sono formulazioni prodotte dalla stessa ditta) LA DIVERSA BIODISPONIBILITA’ PROVOCA PICCHI PLASMATICI DIVERSI SIA IN TERMINI QUANTITATIVI CHE TEMPORALI")
28
Biodisponibilità La biodisponibilità è definita come la frazione di dose di un farmaco (F) che raggiunge la circolazione sistemica e la velocità con cui questo avviene F = Quantità di farmaco nella circolazione sistemica/ Dose somministrata Poiché l’area sotto la curva “concentrazione plasmatica vs. tempo” (AUC) è una misura della concentrazione del farmaco nella circolazione sistemica, la biodisponibilità è di solito definita come : F = AUC/Dose
 è una misura della concentrazione del farmaco nella circolazione sistemica, la biodisponibilità è di solito definita come : F = AUC/Dose.")
29
DETERMINAZIONE DELLA BIODISPONIBILITA’ Si confrontano i livelli plasmatici di un farmaco dopo somministrazione (es: os) con i livelli plasmatici che si ottengono con la somministrazione endovenosa, mediante la quale passa in circolo la totalità del farmaco.
 con i livelli plasmatici che si ottengono con la somministrazione endovenosa, mediante la quale passa in circolo la totalità del farmaco.")
30
DISTRIBUZIONE DEI FARMACI Processo mediante il quale un farmaco passa da un distretto corporeo all’altro fino a raggiungere il sito d’azione Processo di ripartizione in tre fasi liquide: - Plasma - Fluidi extracellulari - Fluidi intracellulari Il plasma rappresenta circa il 4.5% del peso corporeo. I fluidi extracellulari comprendono: fluido interstiziale (16%) e linfa (1.2%). I fluidi intracellulari (30-40%) sono la somma di tutti i contenuti fluidi delle cellule. I fluidi transcellulari (2.5%) comprendono i fluidi cerebrospinale, intraoculare, peritoneale, pleurale e sinoviale
 e linfa (1.2%). I fluidi intracellulari (30-40%) sono la somma di tutti i contenuti fluidi delle cellule. I fluidi transcellulari (2.5%) comprendono i fluidi cerebrospinale, intraoculare, peritoneale, pleurale e sinoviale.")
31
Fattori che influenzano la distribuzione di un farmaco Caratteristiche fisico-chimiche del farmaco Legame della molecola alle proteine plasmatiche (protein binding) Irrorazione degli organi Affinità specifica dei tessuti
 Irrorazione degli organi Affinità specifica dei tessuti")
32
Farmaco idrosolubile Farmaco liposolubile Alla somministrazione Plasma Cellule Plasma All’equilibrio Cellule

33
Legame alle proteine (protein binding) Soprattutto alle albumine Il farmaco legato non attraversa le membrane Equilibrio dinamico tra parte libera e legata Le molecole di farmaco esistono in forma legata o libera in ciascun compartimento, ma solo le molecole libere possono muoversi tra compartimenti
 Soprattutto alle albumine Il farmaco legato non attraversa le membrane Equilibrio dinamico tra parte libera e legata Le molecole di farmaco esistono in forma legata o libera in ciascun compartimento, ma solo le molecole libere possono muoversi tra compartimenti")
34
L’albumina plasmatica è la più importante proteina coinvolta in questi processi di binding ai farmaci; anche -globulina e -glicoproteina legano alcuni farmaci. PlasmaAcqua Extracellulare Proteine Plasmatiche Proteine Tissutali Drug Albumina -------------------- Acidi -glicoproteina acida ----- Basi Globuline -------------------- Steroidi

35
Un complesso proteina-farmaco può causare fenomeni di accumulo per l’elevata affinità del farmaco ad un tessuto, ad esempio al tessuto lipidico quando a.el farmaco è molto lipofilo (anestetici locali) b.il farmaco è abbastanza lipofilo ed è somministrato cronicamente (benzodiazepine) perché il farmaco si lega ai costituenti di un tessuto (tetracicline) Un complesso proteina-farmaco rallenta il metabolismo e l’eliminazione del farmaco. La competizione tra farmaci per binding alle proteine può causare interazioni clinicamente rilevanti.

36
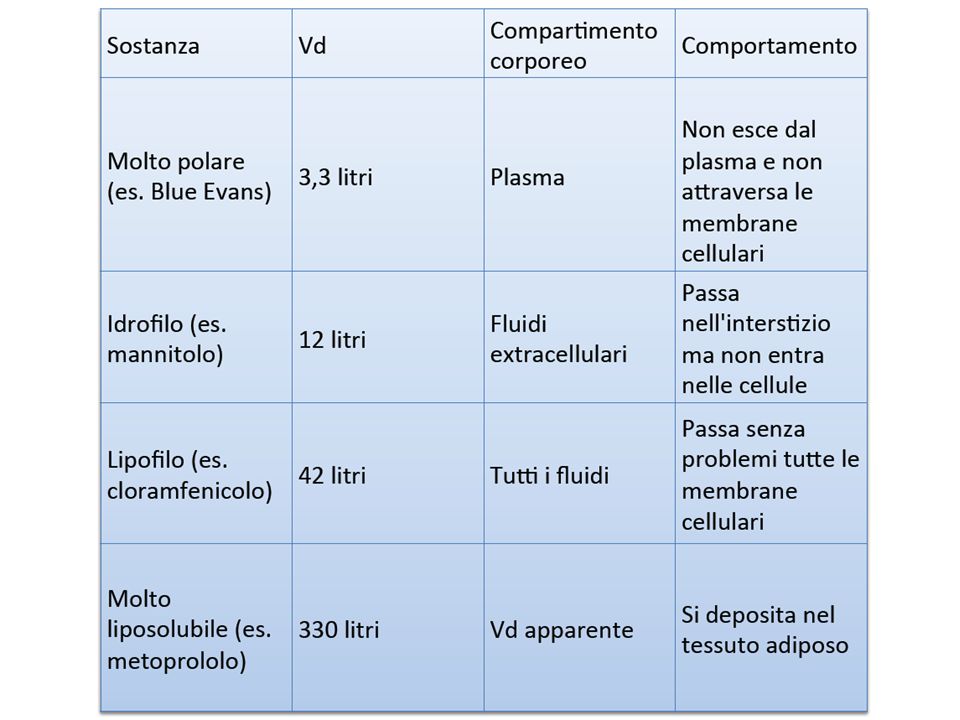
Volume di fluido richiesto per contenere la quantità totale, Q, di farmaco nel corpo alla stessa concentrazione presente nel plasma, Cp Vd = Q/C p Volume di distribuzione PARAMETRI FARMACOCINETICI

37
Indica la distribuzione del farmaco nell’organismo o Più la concentrazione plasmatica di un farmaco è elevata, più il valore numerico del Vd sarà piccolo, ad indicare che il farmaco è scarsamente distribuito o Al contrario, una bassa concentrazione plasmatica rispetto alla dose indicherà che il farmaco si è distribuito in altri distretti dell’organismo ed avrà quindi un elevato volume di distribuzione È influenzato dalla capacità di attraversare le membrane biologiche Grado di ionizzazione Liposolubilità Peso molecolare Volume di distribuzione

39
39 METABOLISMO DEI FARMACI Il metabolismo è l’insieme di trasformazione chimiche che il farmaco subisce all’interno dell’organismo = BIOTRASFORMAZIONE Questa trasformazione ha come scopo la conversione delle sostanze in composti più polari e di norma più idrosolubili, aumentandone la facilità di escrezione

40
40 DOVE AVVIENE LA BIOTRASFORMAZIONE? Sebbene ogni tessuto sia dotato di una certa capacità di metabolizzare i farmaci, il FEGATO è la sede principale del metabolismo Anche altri tessuti come POLMONI, INTESTINO e RENE hanno una attività metabolizzante significativa.

41
Reazioni di fase I Le reazioni di fase I o di FUNZIONALIZZAZIONE hanno la finalità di inserire o mettere in evidenza nella molecola gruppi funzionali come -OH, - NH 2, -COOH OSSIDAZIONE: idrossilazione aromatica idrossilazione in catena laterale deaminazione N-ossidazione N-idrossilazione RIDUZIONE: azoriduzione nitroriduzione alcool deidrogenasi glutatione reduttasi IDROLISI: idrolisi di esteri idrolisi di amidi I farmaci più liposolubili saranno più soggetti a questo tipo di reazioni

42
42 REAZIONI DI FASE I Le reazioni di fase I dipendono, fondamentalmente, da una catena enzimatica di trasporto di elettroni che ha come terminale il citocromo P450 (CYP450) Il reticolo endoplasmatico liscio a livello epatico e/o in tessuti extraepatici (tratto gastrointestinale renale, polmonare, cute e SNC) contiene un importante gruppo di enzimi ossidativi noti come “Ossidasi a funzione mista” o “mono-ossigenasi” i quali richiedono: NADPH O2
 Il reticolo endoplasmatico liscio a livello epatico e/o in tessuti extraepatici (tratto gastrointestinale renale, polmonare, cute e SNC) contiene un importante gruppo di enzimi ossidativi noti come Ossidasi a funzione mista o mono-ossigenasi i quali richiedono: NADPH O2")
43
Citocromo P450 METABOLISMO OSSIDATIVO Il P450 contenente l’ione ferrico (Fe3+) si lega al farmaco (DH), riceve un elettrone dall’enzima NADPH-P450 riduttasi che riduce il Ferro a Fe2+, si combina con l’ossigeno molecolare, un protone e un secondo elettrone per formare il complesso Fe2+OOH-DH. Il gruppo (FeO)3+ estrae un atomo di idrogeno da DH con formazione di un paio di radicali liberi dalla breve durata, liberazione dal complesso del farmaco ossidato (DOH) e rigenerazione dell’enzima P450.
3+ estrae un atomo di idrogeno da DH con formazione di un paio di radicali liberi dalla breve durata, liberazione dal complesso del farmaco ossidato (DOH) e rigenerazione dell’enzima P450..")
44
Reazioni di fase I Ossidazioni Ossidazioni al carbonio Carbonio sp3 Ossidazione di catene alifatiche

45
N,O,S- Dealchilazione

46
Ibuprofen Metabolita principale

47
Esempi

48
Deamminazione ossidativa

49
Carbonio sp2

50
NIH rearrangement

51
Ossidazioni di molecole contenenti N

52
Ossidazioni di molecole contenenti S CLORPROMAZINA Neuroleptico

53
Riduzioni DANTROLENE ipertermia maligna

54
Idrolisi PROCAINAMMIDE Aritmie PROCAINA Anestetico

55
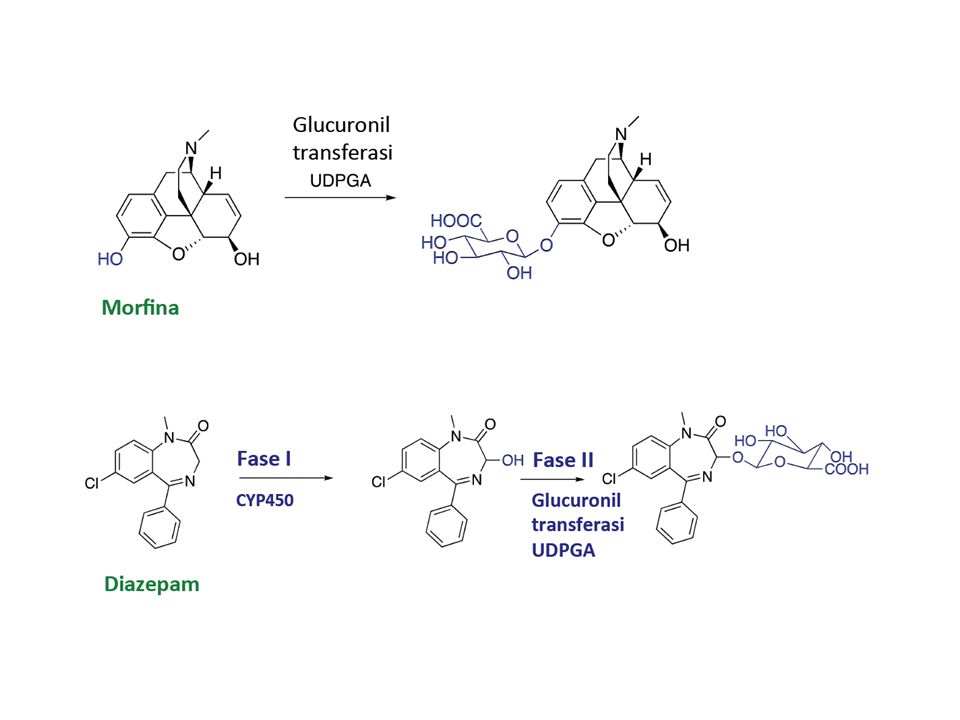
REAZIONI DI FASE II Le reazioni di fase II o di CONIUGAZIONE sono reazioni enzimatiche di biosintesi per mezzo delle quali un composto esogeno o un metabolita derivato dalle reazioni di fase I si lega in modo covalente con una molecola endogena. Reazioni di sintesi CONIUGAZIONE a glucuronide esteri glucuronidici amidi glucuronidiche CONIUGAZIONE A PEPTIDE reazione con glicina taurina METILAZIONE N-metilazione O-metilazione ACETILAZIONE CONIUGAZIONE A SOLFATO In generale i coniugati sono molecole polari facilmente eliminabili.

56
Glucuronidazione

58
Coniugazioni con solfati

59
Coniugazioni con glutatione

60
Coniugazioni con aminoacidi

61
Metilazione

62
Acetilazioni

63
63 METABOLISMO DEI FARMACI La biotrasformazione di un farmaco può portare alla formazione di: inattivi metaboliti inattivi attivi uguale metaboliti attivi dotati di spettro farmacologico uguale a quello del composto d’origine attividiverso metaboliti attivi dotati di spettro farmacologico diverso da quello del composto di origine tossici metaboliti tossici OGNI FARMACO PUO’ DARE ORIGINE A PIU’ DI UN METABOLITA

64
64 Metaboliti attivi dotati di spettro farmacologico uguale a quello del composto d’origine: l’esempio del diazepam La benzodiazepina DIAZEPAM genera due metaboliti dotati della stessa attività farmacologica: OXAZEPAM e NORDIAZEPAM. L’emivita dell’oxazepam è circa ¼ di quella del diazepam L’emivita del nordiazepam è circa il doppio di quella del diazepam. La durata d’azione del diazepam dipende quindi da quale dei due composti viene generato dal corredo di enzimi del paziente

65
65 Metaboliti tossici: l’esempio del paracetamolo PARACETAMOLO Il PARACETAMOLO è un farmaco antiinfiammatorio che viene biotrasformato in un metabolita tossico, il parabenzochinone. Se il paracetamolo viene somministrato a dosi terapeutiche il metabolita tossico viene coniugato con il glutatione ed eliminato. Se il paracetamolo viene somministrato a dosi troppo elevate, il metabolita tossico, dopo aver saturato tutto il glutatione disponibile, si lega alle proteine degli epatociti e causa epatotossicità

66
Apertura dell’epossido da parte di altri nucleofili

67
67 EFFETTO DI PRIMO PASSAGGIO o ELIMINAZIONE (metabolismo) PRESISTEMICO PROCESSO ATTRAVERSO IL QUALE SI HA LA RIMOZIONE DEL FARMACO AD OPERA DEL FEGATO Dopo somministrazione orale molti farmaci sono assorbiti dall’intestino tenue e subito trasportati attraverso la vena porta al fegato dove subiscono una notevole metabolizzazione L’effetto di primo passaggio può limitare così notevolmente la biodisponibilità di un farmaco somministrato per via orale da costringere ad impiegare altre vie di somministrazione Es: gli anestetici locali se somministrati per os vengono metabolizzati ed inattivati in sede epatica. L’uso di questi farmaci prevede solo somministrazione parenterali che permette di evitare il filtro epatico prima di raggiungere la circolazione sistemica

68
FATTORI CHE MODIFICANO IL METABOLISMO DI UN FARMACO FATTORI LEGATI AL FARMACO Liposolubilità e idrosolubilità FATTORI NON LEGATI AL FARMACO Sesso: il metabolismo è maggiore nel maschio che non nella femmina Patologie concomitanti Età Assunzioni di altri farmaci (induttori e inibitori del P450) Cibo: la carne alla brace attiva il sistema enzimatico microsomiale epatico causando aumento del metabolismo della teofillina Variabilità genetica (Polimorfismo genico)
 Cibo: la carne alla brace attiva il sistema enzimatico microsomiale epatico causando aumento del metabolismo della teofillina Variabilità genetica (Polimorfismo genico)")
69
69 ESCREZIONE DEI FARMACI il processo per mezzo del quale un farmaco viene eliminato dall’organismo L’escrezione può avvenire attraverso i reni con l’urina attraverso il dotto biliare e l’intestino con le feci Meno importanti sono l’eliminazione per via polmonare (anestetici generali volatili) e quella attraverso la pelle Etere etilico e stricnina sono esempi di farmaci rapidamente eliminati attraverso l’urina senza andare incontro a fenomeni di accumulo o a trasformazioni metaboliche
 e quella attraverso la pelle Etere etilico e stricnina sono esempi di farmaci rapidamente eliminati attraverso l’urina senza andare incontro a fenomeni di accumulo o a trasformazioni metaboliche")
70
ESCREZIONE DEI FARMACI ESPRESSA COME CLEARANCE PLASMATICA CLEARANCE PLASMATICA O TOTALE La CLEARANCE PLASMATICA O TOTALE esprime la capacità complessiva dell’organismo di eliminare irreversibilmente un farmaco ed è data dalla somma della singole clearance degli organi che concorrono all’eliminazione del farmaco (fegato, rene, polmoni, ecc.) CLEARANCE totale = velocità di eliminazione concentrazione plasmatica Velocità di eliminazione= la somma di tutti i processi di metabolismo ed escrezione
 CLEARANCE totale = velocità di eliminazione concentrazione plasmatica Velocità di eliminazione= la somma di tutti i processi di metabolismo ed escrezione")
71
Tempo di emivita (t 1/2 ) Il t1/2 indica il tempo necessario affinché la concentrazione plasmatica di un farmaco si dimezzi (ovvero si riduca del 50%) Il t1/2 non dipende dalla dose somministrata. Il t1/2 di un farmaco è inversamente proporzionale alla sua clearance e direttamente proporzionale al suo volume di distribuzione t1/2= 0,693xVd/ Cl Clearance Eliminazione completa di un farmaco indipendentemente dalla via di somministrazione Cl = volume di liquido depurato dal farmaco nell’unità di tempo (ml/min/kg) Dipende dal t1/2 e dal Vd
 Dipende dal t1/2 e dal Vd.")
72
Emivita di un farmaco *** Sono necessarie 10 emivite per eliminare il 99,9%*** N° di t½ Frazione di farmaco rimanente 0 1 2 3 4 5 6 7 8 9 10 100% 50% 25% 12.5% 6.25% 3.125% 1.56% 0.78% 0.39% 0.195% 0.0975% Necessario per determinare: Quanto tempo è necessario affinché venga raggiunta la condizione di stato stazionario Quanto tempo è necessario affinché l’organismo elimini completamente il farmaco (tempi di sospensione, durata dell’effetto benefico o tossico) Permette di stimare l’intervallo più opportuno di somministrazione di dosaggi ripetuti (Intervalli fra le dose)
 Permette di stimare l’intervallo più opportuno di somministrazione di dosaggi ripetuti (Intervalli fra le dose)")
73
PRINCIPALI PARAMETRI FARMACOCINETICI Cmax: Cmax: concentrazione massima Misura diretta Tmax Tmax: tempo per raggiungere la Cmax Misura diretta AUC: AUC: area sotto la curva AUC = g *h /l F%: F%: biodisponibilità t½: t½: tempo necessario perché la concentrazione plasmatica si riduca della metà t1/2= 0,693xVd/ Cl = h Vd: Vd: volume di distribuzione Vd = dose /C 0 = mg/kg/ g/l= l/kg Cl: clearance (quantità di farmaco eliminata nell’unità di tempo) Cl=Dose/AUC = ( g /kg)/( g*h)/l = l/h/kg
 Cl=Dose/AUC = ( g /kg)/( g*h)/l = l/h/kg")
74
Grafico di base per valutare le implicazioni terapeutiche dei parametri farmacocinetici.

75
Proprietà farmacocinetiche ideali di un farmaco Da una prospettiva di marketing Deve essere efficaci con un dossaggio una volta / die Uno o due dosi dovrebbero essere sicure ed efficace in tutti gli individui Nessuna rettifica di dosaggio dovrebbe essere richiesto con dosi multiple.

76
Una dose dovrebbe dare la concentrazioni plasmatiche coerenti in tutti gli individui (pazienti). Nessun variabilità nel metabolismo L'escrezione dovrebbe avvenire attraverso entrambi i meccanismi renale ed epatico per quelli pazienti con problemi epatici o renali Rapido nella insorgenza di azione Clerance abbastanza alta; il composto viene rimosso dal corpo velocemente se si osservano effetti collaterali spiacevoli. Nessun accumulo Nessuna interazione con farmaci somministrati a causa di Alto Binding Protein Metabolismo (induzione o inibizione) Interferenze con escrezione Proprietà farmacocinetiche ideali di un farmaco Da una prospettiva di clinica
 Interferenze con escrezione Proprietà farmacocinetiche ideali di un farmaco Da una prospettiva di clinica.")
Presentazioni simili