Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
E T Dinamica molecolare per un sistema isolato ensemble microcanonico (NVE) t
 t")
2
La dinamica molecolare permette di effetturare medie temporali, ovvero di stimare in modo simil-“sperimentale” le osservabili macroscopiche

3
Problema! La dinamica molecolare (ovvero la risoluzione numerica delle Eq. di Newton) permette solo di raggiungere scale temporali limitate (ns). Spesso non sono sufficienti per ottenere un sampling completo dello spazio delle fasi non sempre riesco a effettuare delle medie statisticamente significative. q p can Una barriera cinetica potrebbe impedire di visitare una regione cospicua
 permette solo di raggiungere scale temporali limitate (ns). Spesso non sono sufficienti per ottenere un sampling completo dello spazio delle fasi non sempre riesco a effettuare delle medie statisticamente significative. q p can Una barriera cinetica potrebbe impedire di visitare una regione cospicua.")
4
Crystalline Si at T=300 K Se studio le vibrazioni attorno alla posizione di equilibrio di un solido non me ne accorgo: le vibrazioni non sono un “meccanismo cinetico attivato”

5
Crystalline Si at T=300 K

12
In realtà un solido all’equilibrio è sempre caratterizzato da una certa densità di difetti: se avessimo considerato una cella sufficientemente grande e avessimo potuto simulare l’evoluzione su tempi “umani”, avremmo visto la loro comparsa.

13
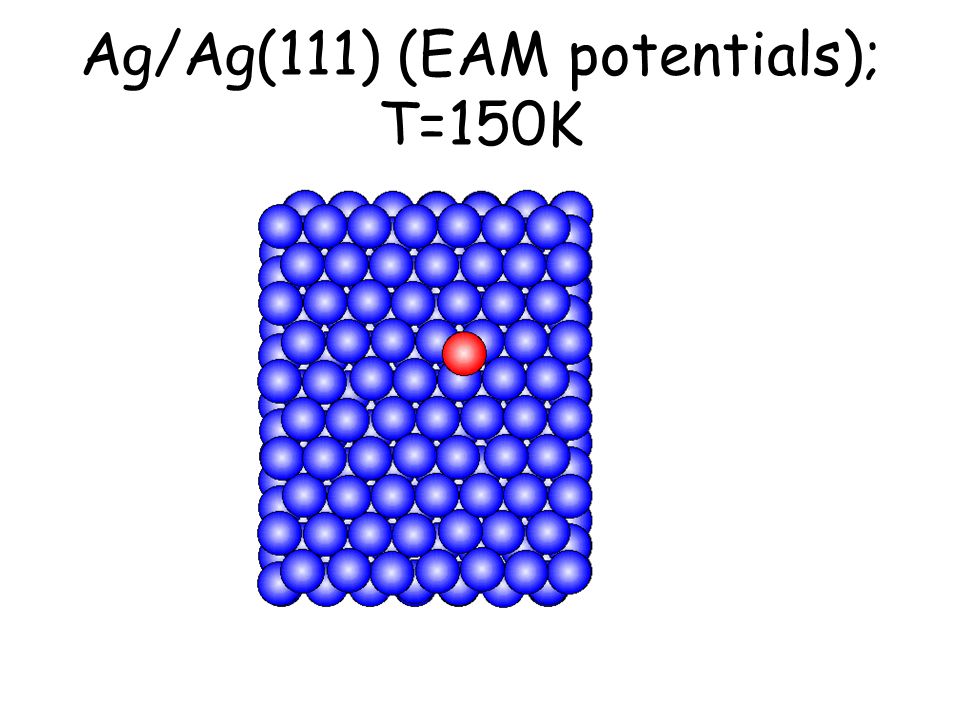
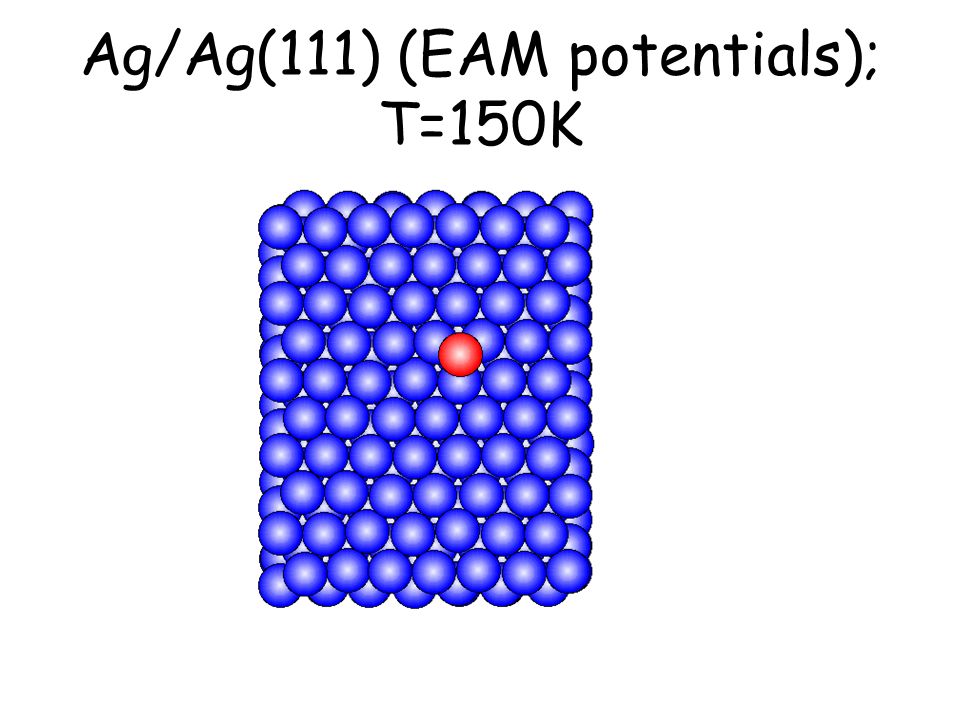
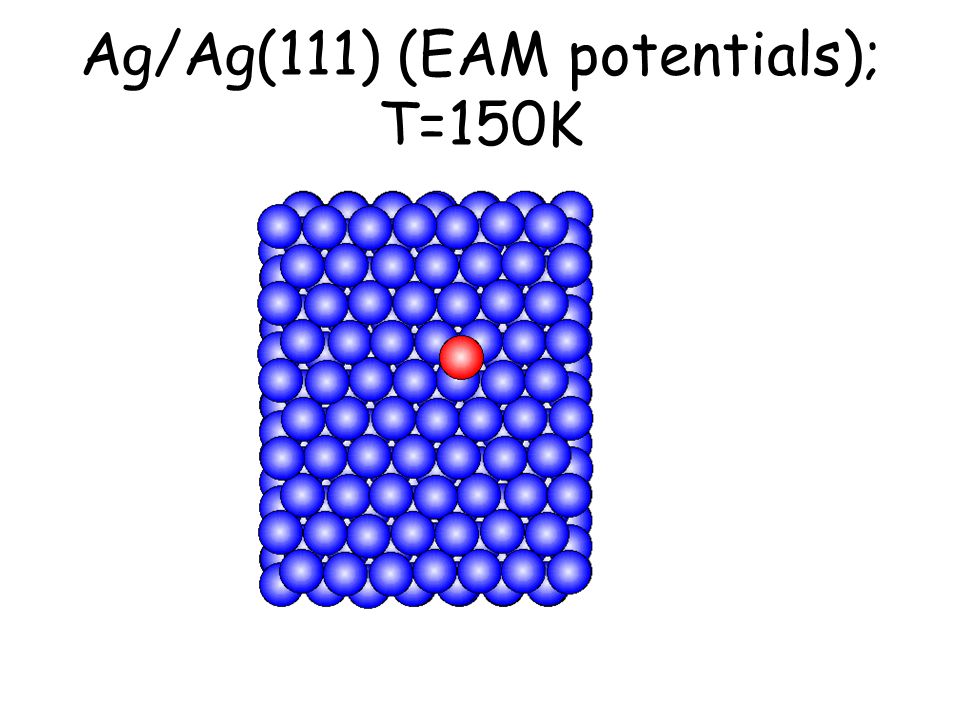
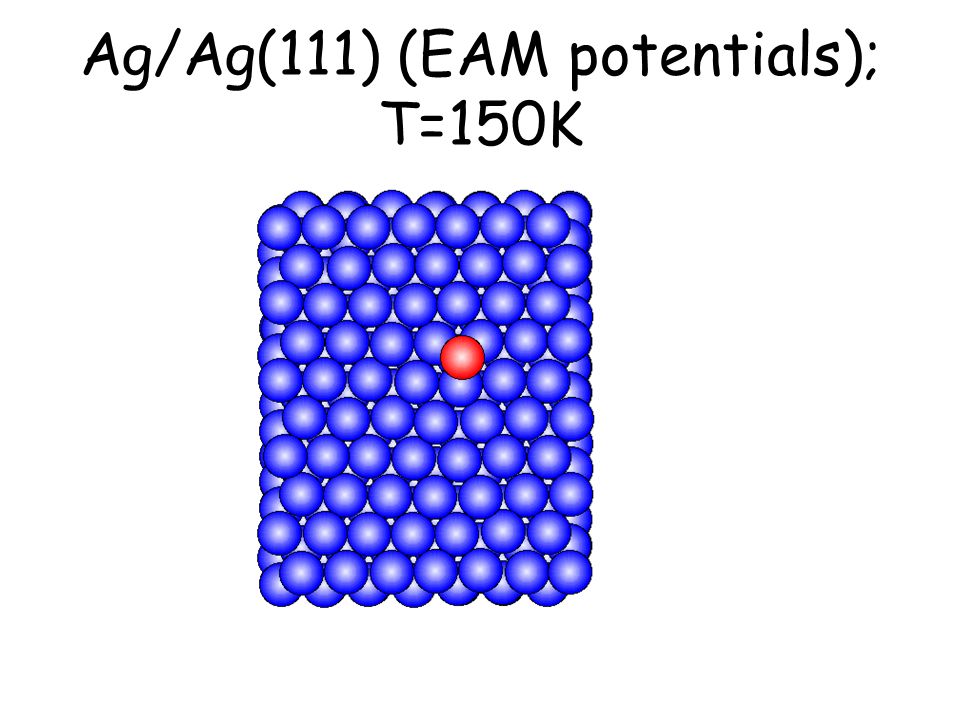
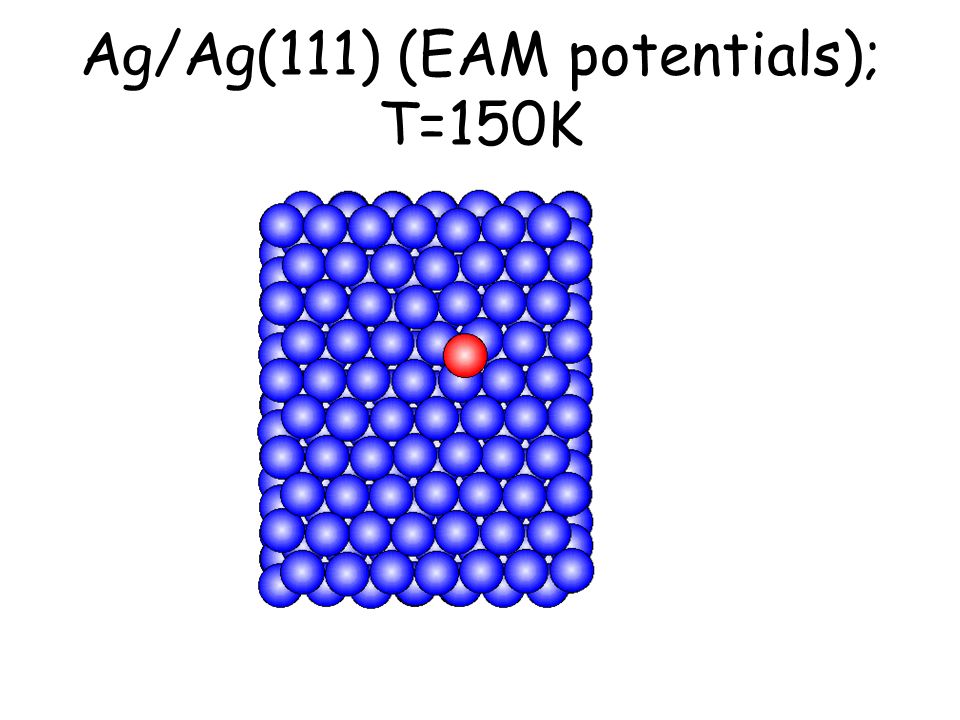
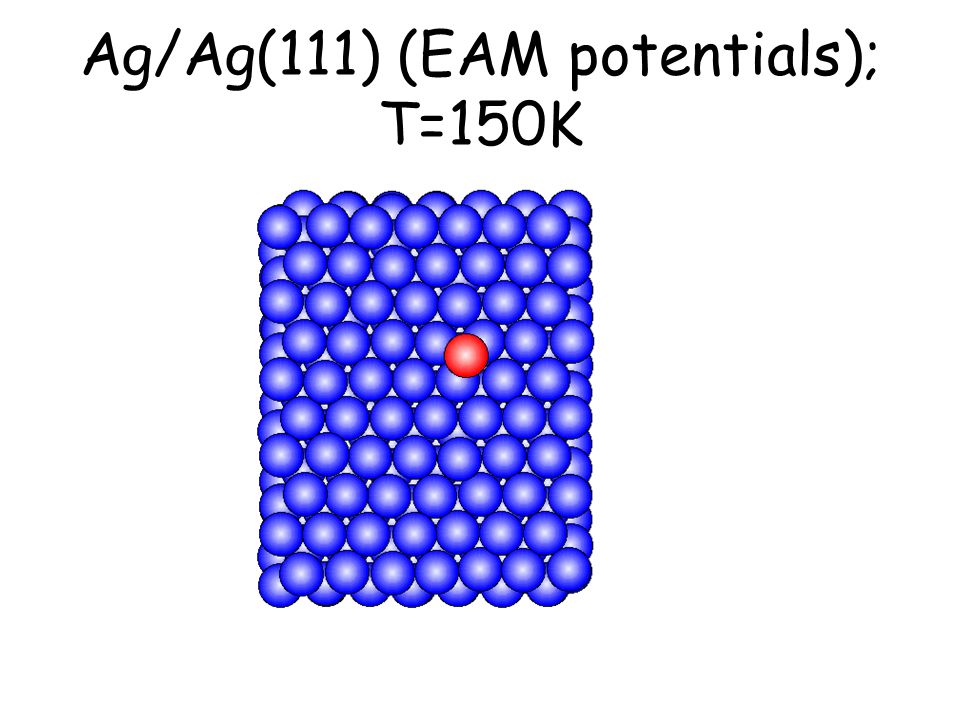
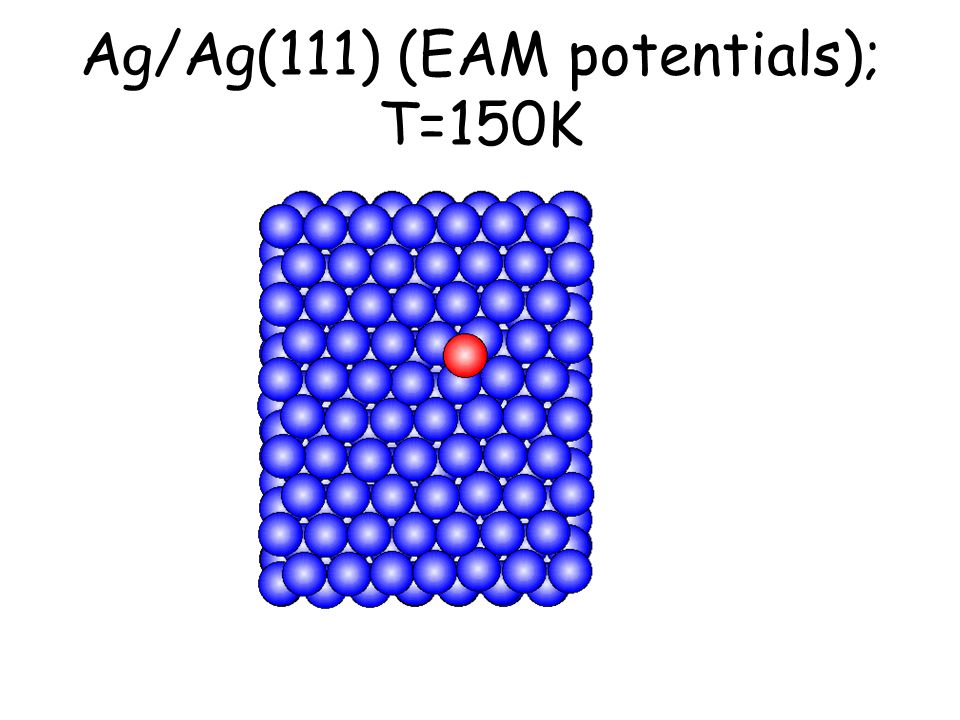
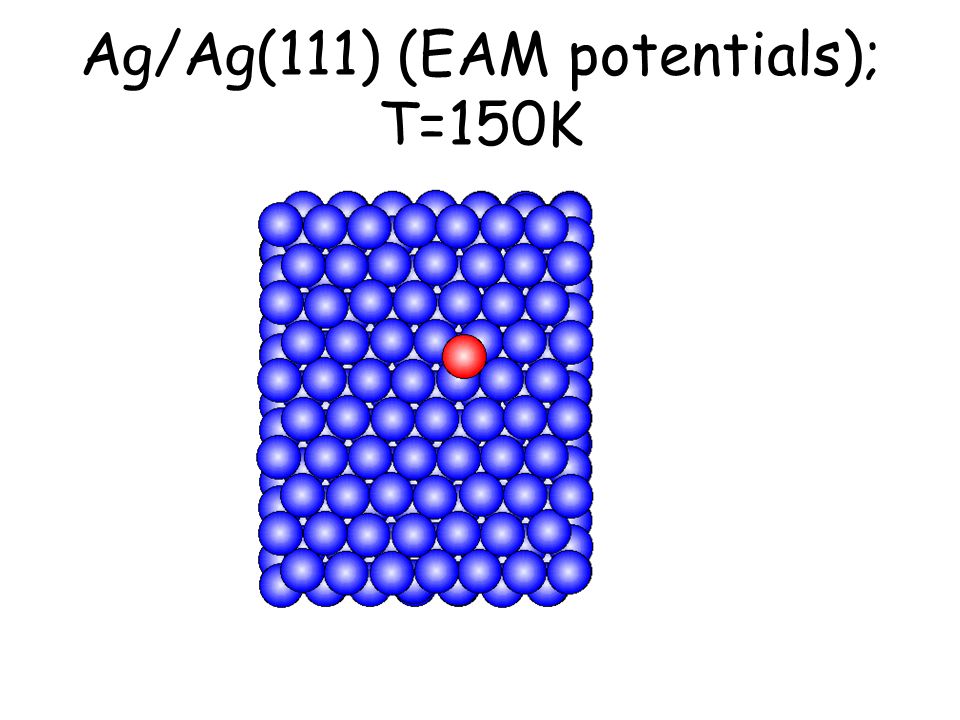
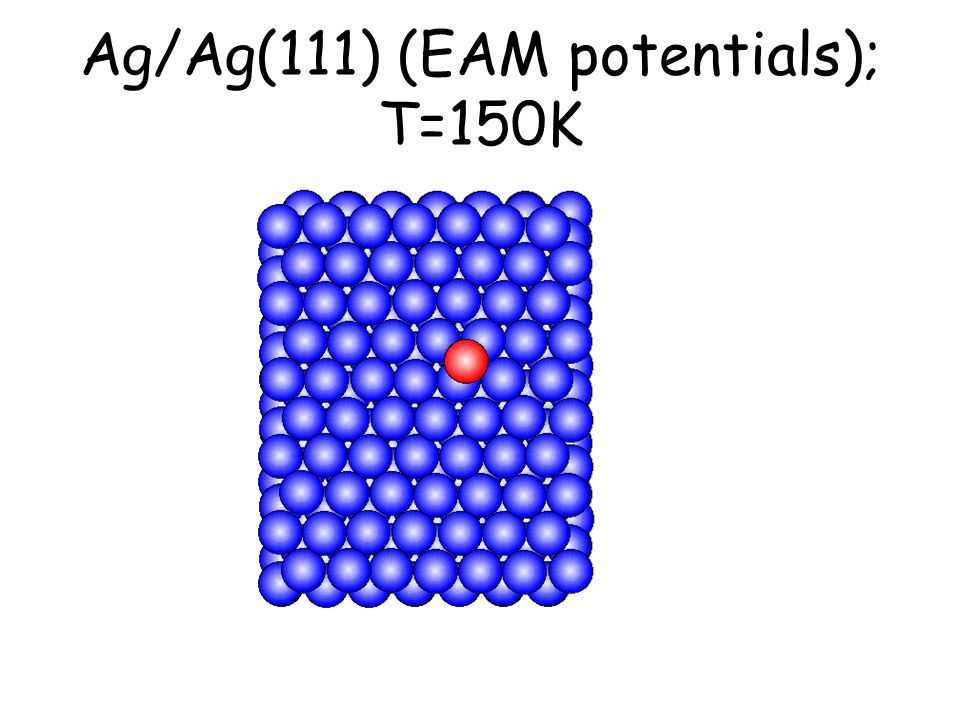
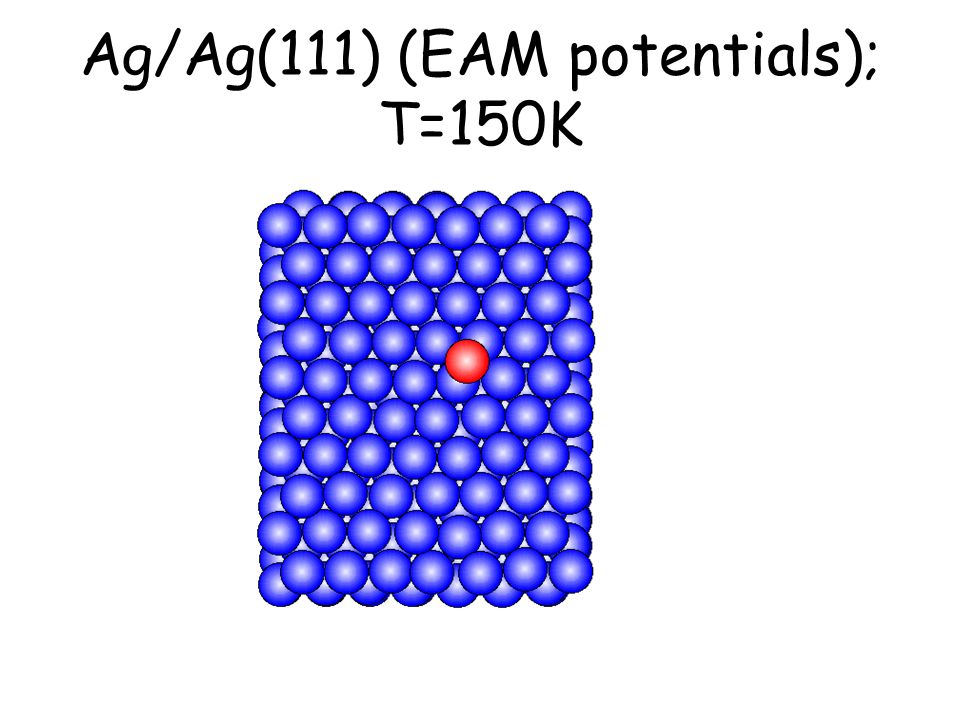
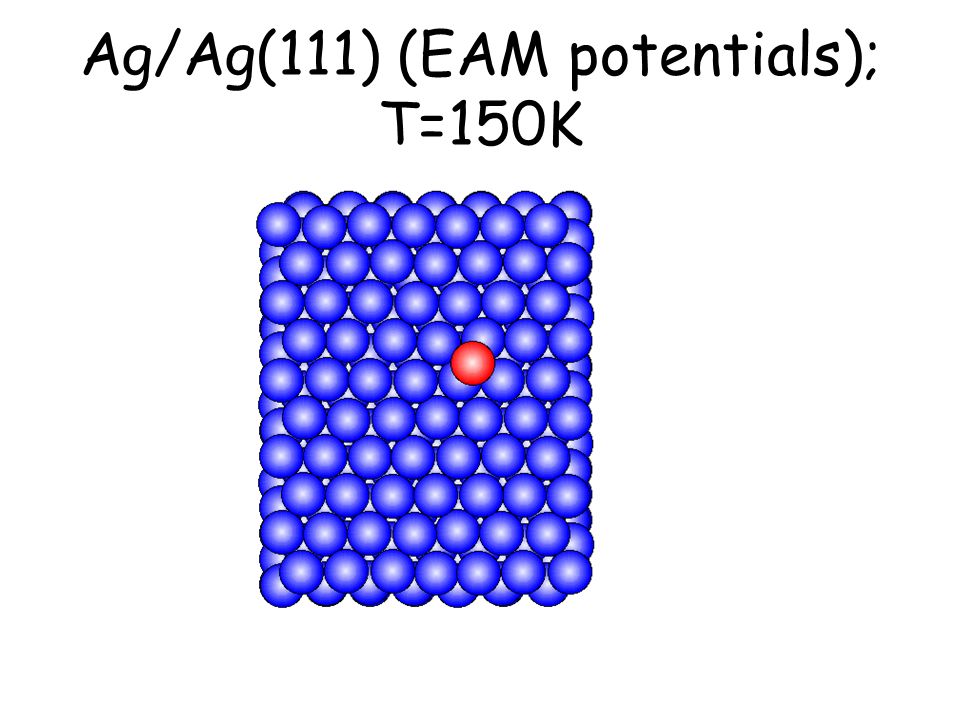
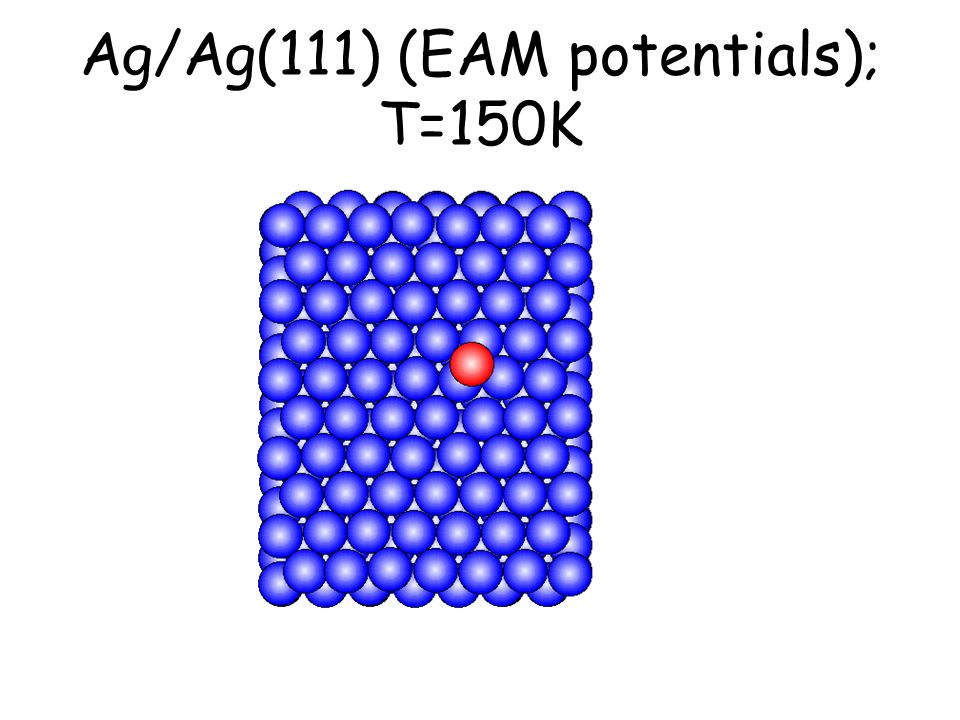
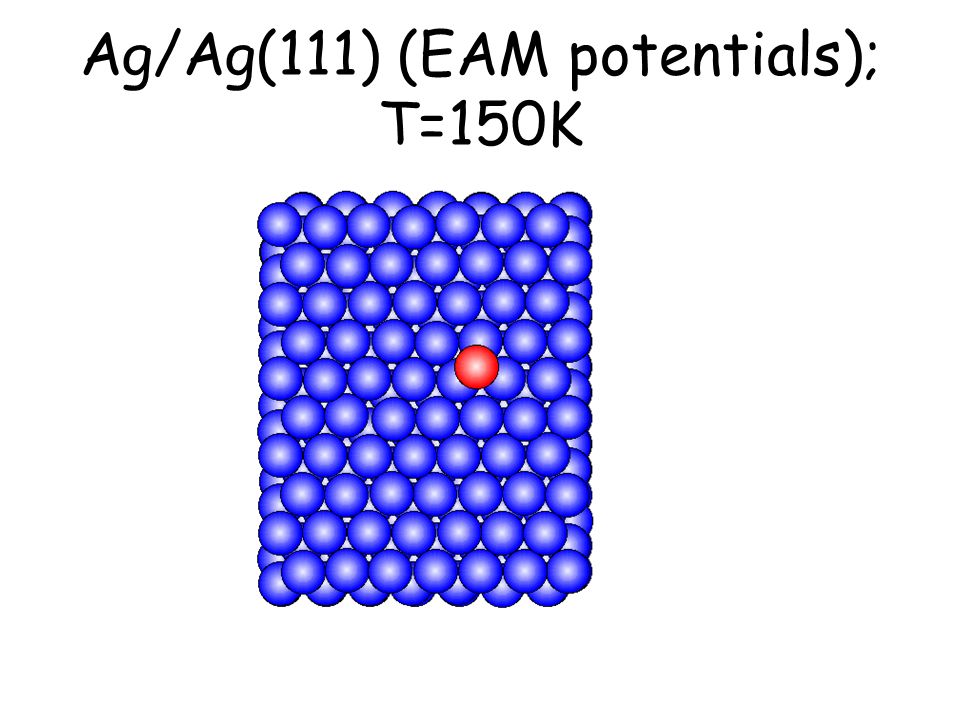
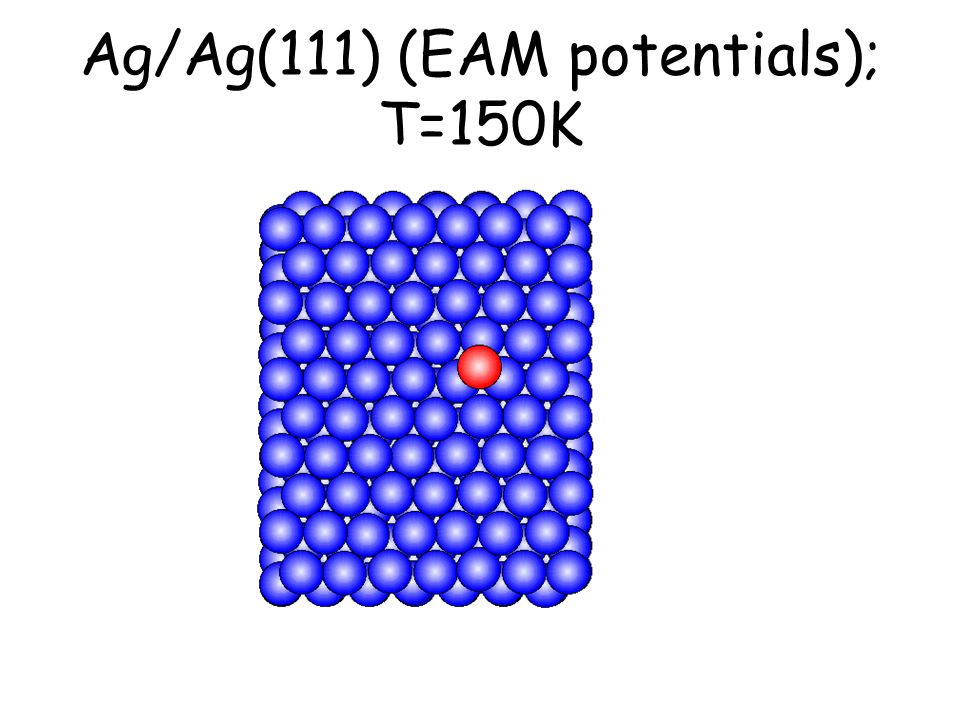
Per osservare meccanismi attivati su scale di tempo accessibili e senza avvicinarsi alla temperatura di melting basta studiare il comportamento di atomi isolati (adatomi) posti su una superficie poco corrugata come la fcc(111). L’evoluzione seguente avviene in pochi ps...

14
Ag/Ag(111) (EAM potentials); T=150K
 (EAM potentials); T=150K")
30
Surface diffusion as a sequence of rare, uncorrelated events Oversimplified picture (1 dimension, 1 particle, “rigid” potential, etc.) The system spends most of its time vibrating around equilibrium position, and it occasionally moves to a new position Time-scale separation (huge at experimental conditions (t D ~1s), negligible at high temperatures, where the picture does not hold.) t vib ~1ps tDtD
 The system spends most of its time vibrating around equilibrium position, and it occasionally moves to a new position Time-scale separation (huge at experimental conditions (t D ~1s), negligible at high temperatures, where the picture does not hold.) t vib ~1ps tDtD")
31
It is obvious that we have a very big problem: in a limited number of steps I could see only “boring vibrations” TimeSteps 1ps1000 1ns1000000 1s1s 1000000000 1ms1000000000000 ab initio MD classical MD real life!

32
Transition State Theory -x x x0x0 x x0x0 State A The rate of escape from A is given by the (equilibrium ensemble average of the) flux exiting through the boundary to state A. System at equilibrium

33
0 E 1D + harmonic approximation for the potential energy: A B

34
Harmonic Transition State Theory 0 frequency prefactor (inverse of typical vibrational time); E = energy of the saddle-point separating state A and state B E is the “activation energy” (or “diffusion barrier”) for the event that causes the system to move from A to B. Arrhenius relation I go from one state to another following the minimum energy path. Along the “reaction coordinate” the energy must raise before diminishing. If I follow the MEP, in all other directions the energy is a minimum. This gives a multidimensional SADDLE POINT

35
Usually very good approximation at experimental conditions, i.e. when :

36
Example: Ag on Ag(111) at T=100K how do we compute E ? often ~10 13 s -1
 at T=100K how do we compute E often ~10 13 s -1")
37
Even using hTST, in order to study the dynamic evolution of a system on a long time scale I need to know all relevant mechanism and to quantify their rates. This is not easy at all. If I’m not interested in the actual dynamics but simply want to estimate equilibrium averages, how can I do?

38
Invece di fare medie temporali, torno a considerare le espressioni d’ensemble. Quelle nell’ensemble canonico sono particolarmente semplici da scrivere... l’integrale a denominatore è 6N-dimensionale.

39
Medie canoniche Chiaramente, nemmeno con i computers posso pensare di risolvere esattamente gli integrali 6N-dimensionali attraverso semplici “griglie” come potrei fare in 1,2,3 dimensioni. Ho bisogno di basarmi sul concetto di “importance sampling”, ovvero devo trovare un trucco per “generare” un numero limitato (fino a 10 8 – 10 9 ce la faccio) di configurazioni che giocano un ruolo chiave nel determinare il valore dell’integrale. Per fortuna c’è il termine esponenziale che garantisce che punti dello spazio delle fasi caratterizzati da grandi valori di H pesano poco …
 di configurazioni che giocano un ruolo chiave nel determinare il valore dell’integrale. Per fortuna c’è il termine esponenziale che garantisce che punti dello spazio delle fasi caratterizzati da grandi valori di H pesano poco ….")
40
Consideriamo un modello “semplicissimo” di sistema interagente, ovvero un sistema di particelle che possono trovarsi solo in due stati diversi, disposte lungo un reticolo ordinato, e interagenti (con energia di interazione costante) solo coi loro primi vicini. Niente traslazioni, rotazioni, vibrazioni. In una dimensione posso immaginarmi il seguente sistema di spin Ho 2 N configurazioni: se il sistema è grande non posso pensare di enumerarle tutte. In 1D non serve, si trova la soluzione esatta. Basta andare in 2D e le cose si complicano molto (articolo terribile di Onsager, che fornisce una soluzione non del tutto generale). Modello studiatissimo: rottura spontanea di simmetria, sistemi ferromagnetici e antiferromagnetici, possibilità di mappare sul modello di Ising molti sistemi di interesse fisico.
. Modello studiatissimo: rottura spontanea di simmetria, sistemi ferromagnetici e antiferromagnetici, possibilità di mappare sul modello di Ising molti sistemi di interesse fisico..")
41
Il modello di Ising risolto col metodo di Monte Carlo Metropolis. 1) Parto da una certa configurazione e ne calcolo l’energia E
 Parto da una certa configurazione e ne calcolo l’energia E.")
42
Il modello di Ising risolto col metodo di Monte Carlo Metropolis. 2) Scelgo uno spin a caso e lo “flippo”; calcolo la nuova energia E’; E=E’-E
 Scelgo uno spin a caso e lo flippo ; calcolo la nuova energia E’; E=E’-E.")
43
Il modello di Ising risolto col metodo di Monte Carlo Metropolis. 3a) Se l’energia è minore “accetto” la nuova configurazione e ripeto l’operazione sulla nuova configurazione E’; E=E’-E <0 accetto Nota: se mi limitassi ad agire in questo modo, starei cercando di minimizzare l’energia del sistema. Ma il comportamento del sistema è determinato, a temperatura finita, dall’energia libera E-TS …
 Se l’energia è minore accetto la nuova configurazione e ripeto l’operazione sulla nuova configurazione E’; E=E’-E <0 accetto Nota: se mi limitassi ad agire in questo modo, starei cercando di minimizzare l’energia del sistema. Ma il comportamento del sistema è determinato, a temperatura finita, dall’energia libera E-TS ….")
44
Il modello di Ising risolto col metodo di Monte Carlo Metropolis. 3a) Se l’energia è maggiore “accetto” la nuova configurazione con probabilità determinata dal fattore alla Boltzmann E’; E=E’-E >0 estraggo un numero casuale p tra 0 e 1; p<exp(- E/kT) accetto, altrimenti rifiuto, riparto dalla configurazione vecchia e riprovo
 Se l’energia è maggiore accetto la nuova configurazione con probabilità determinata dal fattore alla Boltzmann E’; E=E’-E >0 estraggo un numero casuale p tra 0 e 1; p<exp(- E/kT) accetto, altrimenti rifiuto, riparto dalla configurazione vecchia e riprovo.")
45
Soluzione analitica per il modello monodimensionale: m B (B=1 B=1eV)
")
46
Configurational (or equilibrium) Monte Carlo: the Metropolis scheme (not in the more general form) [N. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller, and E. Teller, J. Chem. Phys. 21, 1087 (1953)]
].")
47
1) start from a given configuration x x x2x2 x1x1 x3x3 x4x4
 start from a given configuration x x x2x2 x1x1 x3x3 x4x4")
48
2) define a possible "move" from x x x2x2 x1x1 x3x3 x4x4
 define a possible move from x x x2x2 x1x1 x3x3 x4x4")
49
3) compute the energy difference between the initial and the final state x x2x2 x1x1 x3x3 x4x4
 compute the energy difference between the initial and the final state x x2x2 x1x1 x3x3 x4x4")
50
4) accept the move if E <0 x x2x2 x1x1 x3x3 x4x4
 accept the move if E <0 x x2x2 x1x1 x3x3 x4x4")
51
5) accept the move with probability exp(- E /kT) if E >0; otherwise reject; iterate x x2x2 x1x1 x3x3 x4x4
 accept the move with probability exp(- E /kT) if E >0; otherwise reject; iterate x x2x2 x1x1 x3x3 x4x4")
52
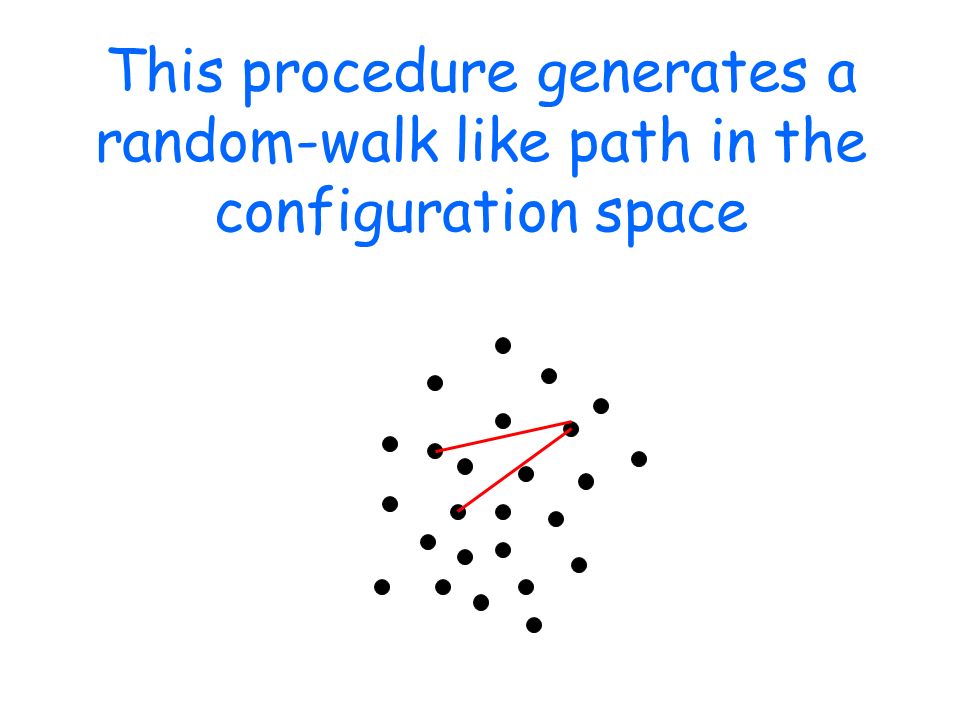
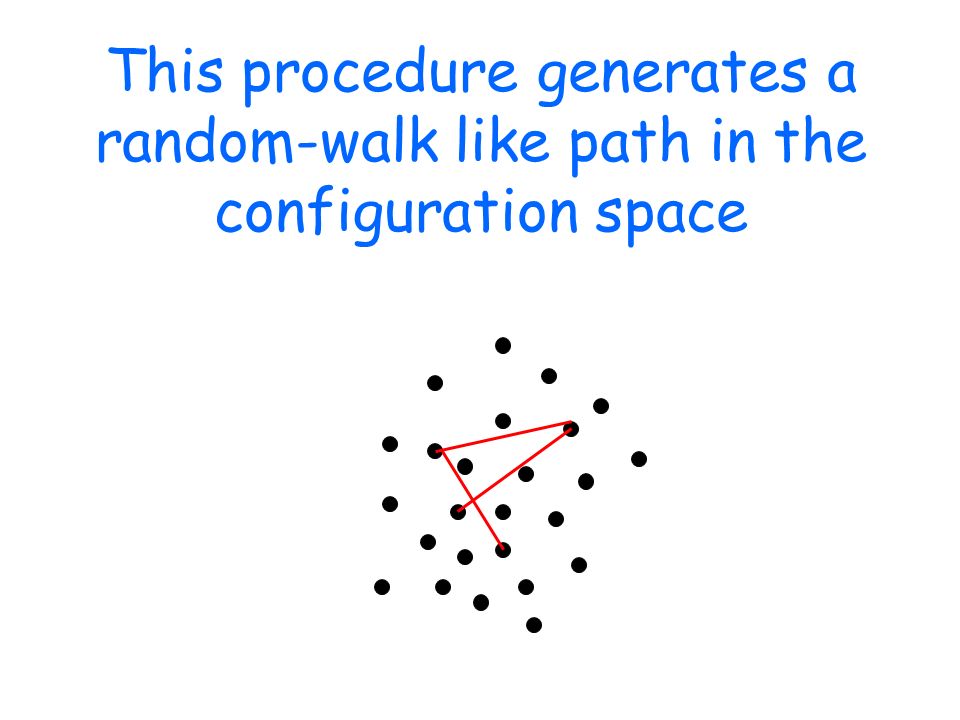
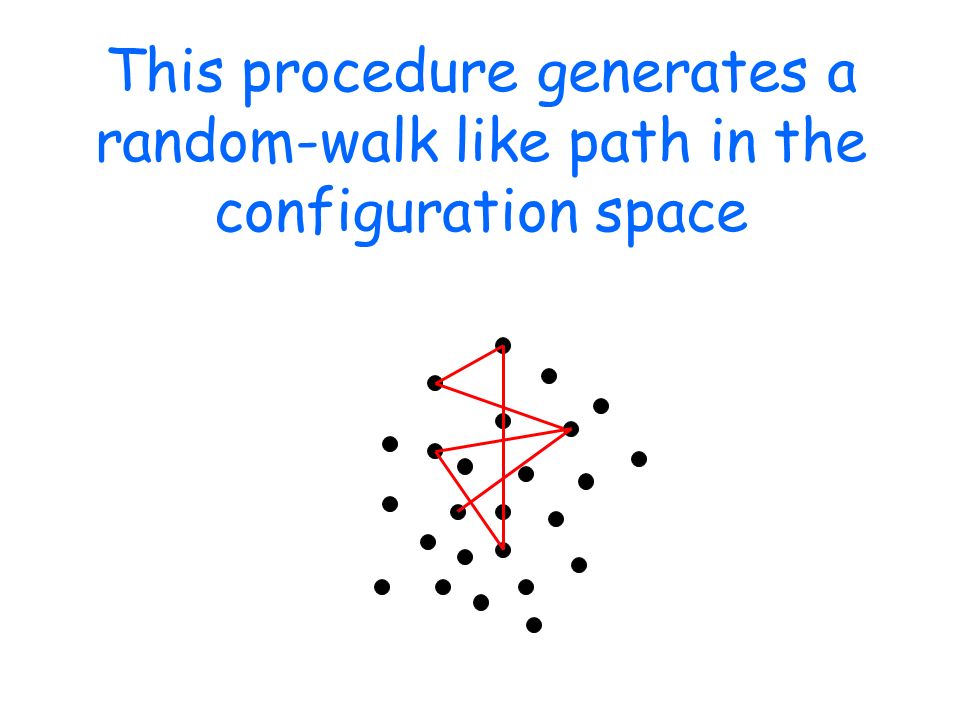
This procedure generates a random-walk like path in the configuration space

56
After a "thermalization" phase, the system equilibrates: the random walk keeps extracting configurations with the correct (canonical) weight
 weight")
57
WARNING: the random walk does not need to be representative of the real system dynamics! (it is not an actual TRAJECTORY; where is the time?)
.")
58
Example: repulsive nn interaction between adsorbates E=0

59
Example: repulsive nn interaction between adsorbates E=J (0.25eV)
")
60
Metropolis move: pick a filled site and an empty one...

61
... and change the occupancy of both of them

62
Half ML coverage; T=50K (~0)
")
68
Since T~0K, no further evolution is possible

69
Half ML coverage; T=2500K

76
Although I'm able to bring the system to equilibrium... I have no information on the time scale since no information on the barriers is available known not known

77
Although I'm able to bring the system to equilibrium... Some of the moves of the configurational random walk are not realistic

78
Il metodo di MC configurazionale (o, alla Metropolis) può essere sfruttato anche per cercare minimi globali. Se il sistema non è banale, in generale occorrerà sfruttare un sampling a temperatura diversa da zero, e non fare confusione tra lo stato di equilibrio del sistema a tale temperatura (minimizza l’energia libera) e lo stato a energia minima.
 e lo stato a energia minima..")
79
Il metodo di MC configurazionale (o, alla Metropolis) può essere sfruttato anche per cercare minimi globali. Se il sistema non è banale, in generale occorrerà sfruttare un sampling a temperatura diversa da zero, e non fare confusione tra lo stato di equilibrio del sistema a tale temperatura (minimizza l’energia libera) e lo stato a energia minima. T=0K; accetto solo mosse in cui l’energia diminuisce; se non parto “vicino” al minimo assoluto potrei NON trovarlo. Possibilità: molte simulazioni indipendenti con posizioni iniziali diverse scelte a caso. Poco efficiente!!!! 1 2 3 1’
 e lo stato a energia minima. T=0K; accetto solo mosse in cui l’energia diminuisce; se non parto vicino al minimo assoluto potrei NON trovarlo. Possibilità: molte simulazioni indipendenti con posizioni iniziali diverse scelte a caso. Poco efficiente!!! ’.")
80
Il metodo di MC configurazionale (o, alla Metropolis) può essere sfruttato anche per cercare minimi globali. Se il sistema non è banale, in generale occorrerà sfruttare un sampling a temperatura diversa da zero, e non fare confusione tra lo stato di equilibrio del sistema a tale temperatura (minimizza l’energia libera) e lo stato a energia minima. kB x T~ E; ho buona probabilità di accettare la mossa in salita, e poi di finire nel minimo più basso 1 2 3 45
 e lo stato a energia minima. kB x T~ E; ho buona probabilità di accettare la mossa in salita, e poi di finire nel minimo più basso")
81
Il metodo di MC configurazionale (o, alla Metropolis) può essere sfruttato anche per cercare minimi globali. Se il sistema non è banale, in generale occorrerà sfruttare un sampling a temperatura diversa da zero, e non fare confusione tra lo stato di equilibrio del sistema a tale temperatura (minimizza l’energia libera) e lo stato a energia minima. Se T è troppo grande non va bene: perdo il peso statistico dato dall’energia più bassa!!!! 1 2 3 4
 e lo stato a energia minima. Se T è troppo grande non va bene: perdo il peso statistico dato dall’energia più bassa!!!")
82
Abbiamo considerato stati discreti. E se fossero continui (sistema di N atomi che interagiscono con potenziali alla Lennard-Jones, liberi di occupare posizioni qualunque)? Basin-hop MC: facciamo diventare gli stati discreti utilizzando lo steepest descent (o altre tecniche di minimizzazione locale)
. Basin-hop MC: facciamo diventare gli stati discreti utilizzando lo steepest descent (o altre tecniche di minimizzazione locale).")
83
Abbiamo considerato stati discreti. E se fossero continui (sistema di N atomi che interagiscono con potenziali alla Lennard-Jones, liberi di occupare posizioni qualunque)? Basin-hop MC: facciamo diventare gli stati discreti utilizzando lo steepest descent (o altre tecniche di minimizzazione locale)
. Basin-hop MC: facciamo diventare gli stati discreti utilizzando lo steepest descent (o altre tecniche di minimizzazione locale).")
84
Abbiamo considerato stati discreti. E se fossero continui (sistema di N atomi che interagiscono con potenziali alla Lennard-Jones, liberi di occupare posizioni qualunque)? Basin-hop MC: facciamo diventare gli stati discreti utilizzando lo steepest descent (o altre tecniche di minimizzazione locale): è come sostituire il vero profilo di energia potenziale con una funzione a gradino.
. Basin-hop MC: facciamo diventare gli stati discreti utilizzando lo steepest descent (o altre tecniche di minimizzazione locale): è come sostituire il vero profilo di energia potenziale con una funzione a gradino..")
86
Configurazione iniziale: ottimizzo tramite steepest descent, salvo posizioni in xold,yold,zold, e calcolo l’energia potenziale Eold

87
Mossa: pesco un atomo a caso iat=rand()*9+1; verificare che non sia 0 o 10, nel caso riestrarre utilizzando un goto o simili.
*9+1; verificare che non sia 0 o 10, nel caso riestrarre utilizzando un goto o simili.")
88
Mossa: cambio la sua x e la sua y a caso ma con delle regole (x es. x=x+4*(2*rand()-1) per muoverlo o a destra o a sinistra al massimo di 4A. Stesso per y)
-1) per muoverlo o a destra o a sinistra al massimo di 4A. Stesso per y).")
89
Sposto e minimizzo tramite steepest; calcolo la nuova energia Eforse. Se Eforse<Eold accetto la mossa, Enew=Eforse, e salvo le nuove posizioni.

90
Se Eforse>Eold e T=0 rifiuto la mossa, Enew=Eold e torno ai vecchi valori di x e y. Se T> 0, calcolo deltaE=Eforse-Eold>0; estraggo un numero a caso aa=rand(). Se aa<exp(-deltaE/kT) accetto la mossa, se no rifiuto.
. Se aa<exp(-deltaE/kT) accetto la mossa, se no rifiuto..")
Presentazioni simili




























 Compri della frutta? Si`, ne compro. Conosci qualche film italiano? Certo, ne.>")


