Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Teoria cinetica dei gas - I

2
Nella teoria cinetica dei gas si estendono i concetti e le leggi della meccanica fino al
livello microscopico, cioè fino a dimensioni atomiche. Vedremo che attraverso una trattazione microscopica è possibile prevedere molte caratteristiche e quantità fisiche osservate a livello macroscopico. Ovviamente, applicando la teoria e le leggi di Newton non ci possiamo aspettare di trattare informazioni sulla posizione e velocità di ogni singola particella di un gas. Adotteremo quindi un approccio statistico, ricavando i valori medi di determinate grandezze. E in effetti, ciò che poi si misura sperimentalmente è il valore medio di alcune grandezze e non le proprietà individuali delle particella di cui si compone il gas.

3
Rivediamo il concetto di equazione di stato di un gas perfetto (o ideale)
Abbiamo visto che l’equazione di stato di una sostanza è la relazione fra la sua pressione, il suo volume specifico e la sua temperatura. Gli esperimenti mostrano che una equazione di stato esiste per qualsiasi sostanza omogenea, sia essa un solido, un liquido o un gas. Sebbene in molti casi non sia possibile formulare in modo semplice l’equazione di stato, una equazione del tipo: F (p, v, T) = 0 esiste per qualsiasi sostanza. Anche se in molti casi noi non siamo in grado di risolvere l’ equazione in questione, la Natura lo sa fare, nel senso che sperimentalmente osserviamo Che le grandezze fisiche in questione obbediscono ad una relazione del genere.
 = 0. esiste per qualsiasi sostanza. Anche se in molti casi noi non siamo in grado di risolvere. l’ equazione in questione, la Natura lo sa fare, nel senso che sperimentalmente osserviamo. Che le grandezze fisiche in questione obbediscono ad una relazione del genere.")
4
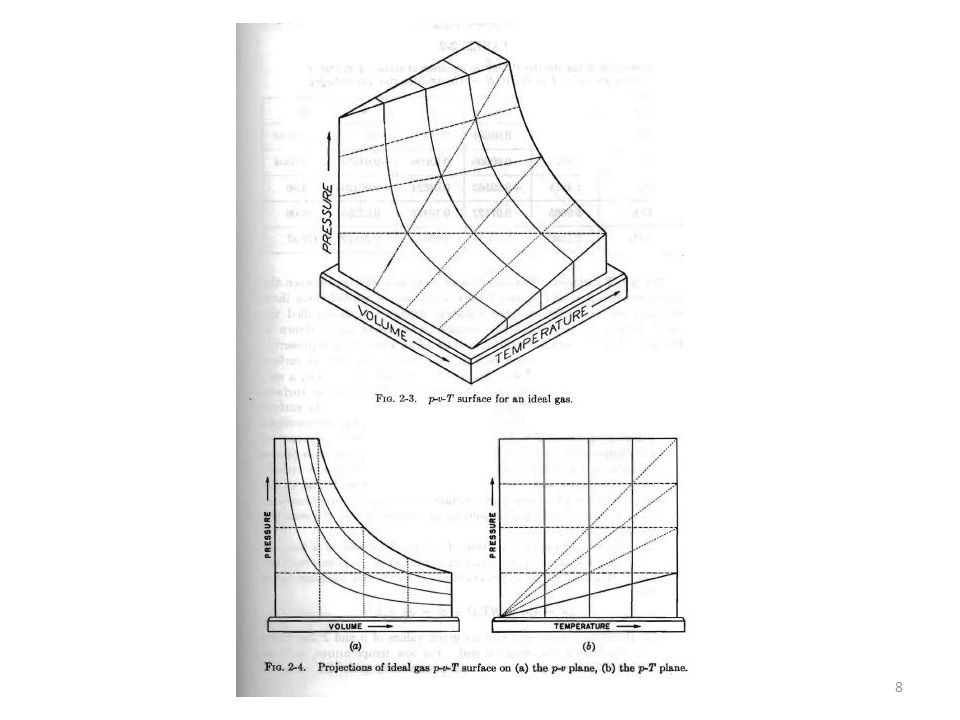
Per avere un’idea di questa tipologia di equazioni ci siamo serviti di grafici in
cui si riportano le misure sperimentali e si interpolano e corrispondenti curve. Abbiamo supposto di avere misurato la pressione p, la temperatura T, il volume V e la massa di un gas, e per il volume abbiamo preso in considerazione il volume specifico v per esempio definito in base al numero di moli n di sostanza , cioè v = V / n Quindi abbiamo considerato tutti i dati raccolti ad un data temperatura T e abbiamo calcolato per ogni set i di misure il rapporto: pi vi /T e abbiamo riportato in un grafico il valore di questo rapporto in funzione della pressione p. Per ogni temperatura T abbiamo ottenuto un set di misure che abbiamo interpolato con una curva continua

5
p v / T T1 T2 T3 R p (atm) E abbiamo fatto una interessante scoperta: ad ogni temperatura i valori della quantità p v / T in funzione della pressione p si dispongono lungo una curva facilmente interpolabile Non solo: le curve convergono tutte verso lo stesso punto R all’avvicinarsi di p a zero E inoltre: si osserva che il valore di p v / T per p 0 è lo stesso per tutti i gas

6
p v / T = R p v = R T o anche: p V = n R T
Utilizzando per la grandezza di p v / T le seguenti unità di misura: p nt / m2 v m3 / kg-mole T °K E’ risultato che: R = x 103 joules / kg-mole-°K La costante R è denominata costante universale dei gas. Risulta quindi che a basse pressioni si può scrivere per tutti i gas: p v / T = R p v = R T o anche: p V = n R T

7
Abbiamo anche visto che in molte applicazioni, risulta conveniente definire un «gas ideale»
che obbedisce alla legge: p v = R T per tutti i valori di pressione. Questa è quella che si chiama equazione di stato di un gas ideale p v / T Qualsiasi T R p (atm)
")
9
Come si può vedere le trasformazioni isoterme, cioè quelle che avvengono a
temperatura T costante, sono rappresentate da iperboli nel piano p-V: p V = n R T Abbiamo anche visto per esempio che la semplicità di questa relazione permette di calcolare il lavoro fatto da un gas ideale durante una espansione isotermica.

10
Passiamo adesso alla definizione di un modello di gas secondo la teoria cinetica
Adotteremo le seguenti esemplificazioni: Un gas consiste di particelle chiamate molecole. A seconda del gas, ciascuna molecola potrà consistere di un atomo o di un gruppo di atomi. In molti casi tutte le molecole saranno considerate identiche. Le molecole sono in moto casuale e obbediscono alle Leggi di Newton. Le molecole si muovono in tutte le direzioni con varie velocità. Nel trattarne il moto assumeremo la validità della meccanica newtoniana. Il numero totale di molecole è grande. Il moto di ogni singola molecola può cambiare bruscamente a seguito di urti. Una qualsiasi molecola seguirà in generale un moto a zig-zag. Tuttavia essendoci un numero elevatissimo di urti supporremo che rimanga inalterata la distribuzione delle velocità molecolari.

11
Il volume delle molecole è una frazione trascurabile del volume del gas. In sostanza,
anche se abbiamo molte molecole, esse sono estremamente piccole. Sappiamo infatti che il volume di un gas può essere facilmente ridotto e quindi questa ipotesi è certamente plausibile. Sulle molecole non agisce alcuna forza apprezzabile, se non durante una collisione. Quindi durante due urti successivi ogni singola molecola si muove di moto uniforme, cioè a velocità costante e in liea retta. Poiché abbiamo supposto che le dimensioni di una molecola siano piccole, la distanza media fra le molecole è relativamente grande, quindi assumeremo che il raggio di azione delle forze molecolari sia confrontabile con le dimensioni molecolari Gli urti sono perfettamente elastici e di durata trascurabile. Cioè gli urti fra le molecole e con le pareti conservano sia l’energia cinetica che la quantità di moto.

12
tutte le ipotesi enunciate siano rigorosamente vere
Nello sviluppo della teoria cinetica, si suppone che in un gas perfetto (o ideale) tutte le ipotesi enunciate siano rigorosamente vere In particolare si suppone di potere trattare gli urti fra le molecole assimilando ad urti fra «biglie completamente elastiche» Per giustificare questa assunzione, è bene entrare un attimo nel merito di come nella teoria cinetica vengono modellate le forze intermolecolari
 tutte le ipotesi enunciate siano rigorosamente vere. In particolare si suppone di potere trattare gli urti fra le molecole assimilando ad urti. fra «biglie completamente elastiche» Per giustificare questa assunzione, è bene entrare un attimo nel merito di come. nella teoria cinetica vengono modellate le forze intermolecolari.")
13
Forze intermolecolari
Le forze fra molecole sono di origine elettromagnetica. Tutte le molecole contengono infatti cariche elettriche in movimento, e sebbene ogni molecola sia elettricamente neutra, le molecole interagiscono fra di loro elettricamente. Possiamo immaginare che quando due molecole si avvicinano l’una all’altra, le cariche di ciascuna sono disturbate, cosi da disporsi su una simmetria differente da quella usuale. Ne risulta una forza intermolecolare. Questo fenomeno si verifica a distanze sufficientemente piccole, e si tratta quindi di forze a corto range Se le molecole si avvicinano molto, così che le cariche esterne cominciano a sovrapporsi, la forza intermolecolare diventa repulsiva.

14
La tipica «buca di potenziale» che caratterizza la forza intermolecolare fra due molecole
appare come in figura: U r Repulsivo Attrattivo

15
Avevamo già visto in meccanica che la presenza di un potenziale repulsivo genera urti del tutto paragonabili agli urti «per contatto» fra delle biglie:

16
In generale, possiamo immaginare che nel caso di un potenziale attrattivo l’effetto
di URTO che si osserva sia simile e cioè del tutto paragonabile al caso di orbite aperte: Orbita chiusa: Orbita aperta:

17
Quindi anche un potenziale attrattivo produce a tutti gli effetti un «urto», del tutto
simile a quello prodotto da un potenziale repulsivo.

18
Quindi a tutti gli effetti, l’esistenza della «buca di potenziale» corrispondente alle
forze intermolecolari , genera una varietà di urti dipendente dal raggio di azione (cioè della sezione d’urto), così come avevamo già visto in meccanica.
, così come avevamo già visto in meccanica.")
19
Sezione d’urto σ

20
Calcolo cinetico della pressione
Consideriamo un gas in un recipiente di forma cubica di lato l, dotato di pareti perfettamente elastiche: y A2 A1 v x z Consideriamo le due facce A1 e A2 ortogonali all’asse x ciascuna di area l 2 e consideriamo una particolare molecola che viaggia a velocità v

21
Δp = pf − pi = −m vx − (m vx ) = −2 m vx
Scomponiamo la velocità v nelle sue componenti vx vy e vz. Se questa particella urta contro la parete A1, rimbalzerà indietro con una componente della velocita vx uguale e opposta, mentre non vi sarà alcun cambiamento in vy e vz . Quindi la variazione Δp di quantità di moto della particella sarà data dalla: Δp = pf − pi = −m vx − (m vx ) = −2 m vx che è una variazione di quantità di moto ortogonale ad A1 Quindi la quantità di moto trasmessa ad A1 sarà: 2 m vx Supponiamo adesso che questa particella raggiunga A2 senza urtare altre particelle. Il tempo impiegato per attraversare il cubo sarà Δt = l / vx
 = −2 m vx. che è una variazione di quantità di moto ortogonale ad A1. Quindi la quantità di moto trasmessa ad A1 sarà: 2 m vx. Supponiamo adesso che questa particella raggiunga A2 senza urtare. altre particelle. Il tempo impiegato per attraversare il cubo sarà. Δt = l / vx.")
22
numero collisioni = vx / 2 l
Di nuovo, urtando A2 la particella inverte la sua velocità e se non subisce altri urti raggiunge nuovamente A1 con la stessa velocità. L’intero percorso richiede un tempo Δt = 2 l / vx quindi il numero di collisioni per unità di tempo che questa particella subisce con la parete A1 è dato da: numero collisioni = vx / 2 l e quindi la quantità di moto che trasmette a A1 nell’unità di tempo è data da: numero collisioni x Δp = ( vx / 2 l ) (2 m vx) = m v2x / l
 (2 m vx) = m v2x / l.")
23
Pressione p = (m / l3 )( v2x1 + v2x2 + ….. v2xN )
Per determinare la forza esercitata su A1 ricordiamo che la forza è proprio la variazione di quantità di moto per unità di tempo: F = Δp /Δt . E poiché abbiamo appena visto che la variazione di quantità di moto per unità di tempo vale: Δp /Δt = m v2x / l. per determinare la forza totale esercitata da tutte le molecole su A1 dobbiamo sommare m v2x / l per tutte le particelle. Poi per trovare la pressione divideremo questa forza totale per la superfice, cioè l2. Cioè: Pressione p = (m / l3 )( v2x1 + v2x2 + ….. v2xN ) Avendo indicato con N il numero totale di particelle, indicheremo con n = N / l3 il numero di particelle per unità di volume e quindi l3 = N / n e quindi risulta: p = m n v2x1 + v2x2 + ….. v2xN 𝑁
( v2x1 + v2x2 + ….. v2xN ) Avendo indicato con N il numero totale di particelle, indicheremo con n = N / l3 il numero. di particelle per unità di volume e quindi l3 = N / n e quindi risulta: p = m n v2x1 + v2x2 + ….. v2xN 𝑁.")
24
p = ρ < v2x > = 1 3 ρ < v2 >
Osserviamo che m n è semplicemente la massa per unità di volume e cioè la densità ρ. Inoltre la quantità v2x1 + v2x2 + ….. v2xN 𝑁 rappresenta il valore medio di v2x per tutte le particelle nel cubo che indicheremo con < v2x > Quindi risulta: p = ρ < v2x > Per ogni particella si ha: v2 = v2x + v2y + v2z . Inoltre, poiché le particelle si muovono a caso si ha: v2x = v2y = v2z e quindi il valore di ognuno è pari a 1/3 del valor medio di v2. Quindi: < v2x > = < v2 > e cioè: p = ρ < v2x > = ρ < v2 >

25
In tutto questo però abbiamo trascurato gli urti fra particelle
In tutto questo però abbiamo trascurato gli urti fra particelle. Tuttavia, trattandosi di urti elastici fra particelle di equale massa, risultano scambi di velocità in cui ci sarà sempre una particella che prosegue il suo moto con la velocità di un’altra particella. Quindi trascurare gli urti non dovrebbe alterare il risultato macrosopico ottenuto. La trattazione fatta si applica ovviamente anche a tutte le pareti del cubo.

26
p = ρ < v2x > = 1 3 ρ < v2 > = 1 3 m n < v2 >
Interpretazione cinetica della temperatura. Indichiamo con M la massa totale del gas in questione. Avremo M = N m . Poiché n = N/V potremo riscrivere l’equazione precedente: p = ρ < v2x > = ρ < v2 > = m n < v2 > come di seguito: p = m n < v2 > = 𝑚 N 𝑉 < v2 > = 1 3 M < v2 > 𝑉 E cioè: p V = M < v2 >

27
1 2 M < v2 > = 3 2 R T p V = 1 3 M < v2 >
Osserviamo con attenzione questa relazione: p V = M < v2 > la quantità a destra è i 2/3 dell’energia cinetica di traslazione delle molecole, cioè possiamo scrivere: p V = x M < v2 > Se supponiamo di avere una sola mole di gas, cosicché si possa sostituire M con M, il peso di una grammo-molecola. Per una mole di gas ideale sappiamo che l’equazione di stato e: p V = R T Confrontando le due relazioni si ha: M < v2 > = R T

28
(Ricordate questo: ½ kT per ogni grado di libertà)
Questa relazione: M < v2 > = R T ci dice che: l’energia cinetica totale di traslazione delle molecole di un gas è direttamente proporzionale alla temperatura T. Si può riscrivere questa equazione per una singola molecola come segue: m < v2 > = k T (Ricordate questo: ½ kT per ogni grado di libertà) dove k è una costante universale denominata costante di Boltzman, dove: k = 𝑅 𝑁/𝑛 con ( N/n ) = numero di molecole / numero di moli = N0 (numero di Avogadro)
 dove k è una costante universale denominata costante di Boltzman, dove: k = 𝑅 𝑁/𝑛 con ( N/n ) = numero di molecole / numero di moli = N0 (numero di Avogadro)")
29
Calore specifico di un gas ideale
Abbiamo descritto le molecole di un gas ideale come delle biglie perfettamente elastiche. Durante gli urti quindi le molecole non rimangono deformate, e poiché gli urti sono istantanei, non c’è nessun accumulo di energia potenziale nelle biglie. Quindi in un gas non c’è energia potenziale interna. L’energia interna è tutta energia di movimento: energia cinetica. Abbiamo visto che l’energia cinetica media per molecola è 3/2 k T così che L’energia interna U di un gas contente N molecole è data da: U = N k T = n R T Cioè: l’energia interna di un gas dipende solo dalla temperatura e non dal volume o dalla pressione.

30
CV = calore specifico molare a volume costante
Il calore specifico di una sostanza è la quantità di calore per unità di massa necessaria per innalzarne la temperatura di 1 °K. L’unità di massa conveniente per un gas può essere certamente la mole. Il calore specifico corrispondente è chiamato calore specifico molare (o capacità termica molare), ed è rappresentato da C. Per un gas, definiremo: CV = calore specifico molare a volume costante Cp = calore specifico molare a pressione contante
, ed è rappresentato da C. Per un gas, definiremo: CV = calore specifico molare a volume costante. Cp = calore specifico molare a pressione contante.")
31
Se i producesse lo stesso innalzamento di temperatura a pressione
Se aumentiamo di dT la temperatura di n moli di un gas a volume costante, il calore ceduto sarà dQ = n CV dT. In generale, in base al I Principio si avrà: dQ = dU + pdV ma poiché la trasformazione è avvenuta a volume costante, dV= 0 pdV= 0 e quindi: dQ = dU n CV dT = dU Se i producesse lo stesso innalzamento di temperatura a pressione costante invece, il gas si espanderebbe e in questo caso fare lavoro esterno pdV, ma il mutamento di energia interna sarebbe sempre lo stesso in quanto come abbiamo visto dipende solo da T

32
Equipartizione dell’energia
Un passo avanti nella modellizzazione di un gas ideale può essere fatto non trattando necessariamente le molecole come masse puntiformi ma immaginando che possano avere una struttura fisica, cioè in sostanza una forma. In questo caso, una molecola potrà anche avere un moto rotatorio attorno ad un asse di simmetria o anche un moto vibratorio. In questo caso, oltre all’energia cinetica di traslazione che esprimeremo con termini del tipo ½ m v2, per ogni molecola potremo supporre l’esistenza di energia rotazionale, che esprimeremo con termini del tipo ½ I ω2 e di energia potenziale vibrazionale, che esprimeremo con termini del tipo ½ k x2. Si dimostra su base statistica che quando il numero di molecole i questione è elevato : Tutti i termini di energia interna hanno lo stesso valore medio, e questo valore medio dipende solo dalla temperatura (Teorema di equipartizione dell’energia)
")
33
Teoria cinetica dei gas - II

34
Cammino libero medio Il cammino percorso da una molecola tra due urti successivi è definito cammino libero. Durante il cammino libero, la molecola si muove con velocità costate lungo una linea retta. La distanza media fra due urti successivi è definita cammino libero medio. Il cammino libero medio dipende da: Le dimensioni fisiche delle molecole La densità di molecole Infatti: se le molecole fossero così grandi e/o così numerose da occupare tutto il volume disponibile, il cammino libero medio sarebbe presoché nullo.

35
d d Per calcolare il cammino libero medio, procederemo così:
Supponiamo che le molecole siano sfere di diametro d. La sezione d’urto per una collisione di contatto sarà πd2. L’urto infatti avviene quando i centri delle due molecole si avvicinano entro una distanza d. d d

36
Un modo semplice per affrontare il problema è quindi quello di considerare una
molecola bersaglio di diametro 2d (raggio d) e tutte le altre puntiformi. Se seguiamo il movimento di una molecola di diametro 2d che si muove a velocità v in un mezzo popolato di molecole puntiformi, troveremo che in tempo Δt descriverà un cilindro avente sezione πd 2 e lunghezza v Δt. πd 2 v Δt In questo intervallo di tempo v Δt la molecola si scontrerà con tutte le molecole i cui centri si presentino nel cilindro.
 e tutte le altre puntiformi. Se seguiamo. il movimento di una molecola di diametro 2d che si muove a velocità v in un mezzo. popolato di molecole puntiformi, troveremo che in tempo Δt descriverà un. cilindro avente sezione πd 2 e lunghezza v Δt. πd 2. v Δt. In questo intervallo di tempo v Δt la molecola si scontrerà con tutte le molecole i cui centri si presentino nel cilindro.")
37
<l> = v Δt n x (πd 2 x v Δt) = 1 n πd 2
Quindi, se ci sono n molecole per unità di volume, il numero di urti nell’intervallo di tempo Δt sarà: numero di urti in Δt = n x (πd 2 x v Δt) Il cammino libero medio è la distanza media <l> fra gli urti (da intendersi come la distanza percorsa fra un urto e il successivo). La distanza media <l> è la distanza media fra gli urti, cioè la distanza totale v Δt percorsa nel tempo Δt divisa per il numero di urti che occorrono nello stesso tempo, ossia: <l> = v Δt n x (πd 2 x v Δt) = 1 n πd 2
 Il cammino libero medio è la distanza media <l> fra gli urti (da intendersi come la. distanza percorsa fra un urto e il successivo). La distanza media <l> è la distanza. media fra gli urti, cioè la distanza totale v Δt percorsa nel tempo Δt divisa per il. numero di urti che occorrono nello stesso tempo, ossia: <l> = v Δt n x (πd 2 x v Δt) = 1 n πd 2.")
38
Questo in realtà è un risultato approssimato in quanto è stato ricavato nell’ipotesi che la molecola incontri dei «bersagli fissi». In realtà i bersagli sono in moto. Ne segue che la frequenza di urti è leggermente superiore, e si dimostra che la formula corretta è: <l> = n πd 2

39
Distribuzione delle velocità molecolari
Fino adesso abbiamo discusso in termini di velocità quadratica media delle molecole. In realtà ogni molecola, pur mantenendo questa velocità quadratica media, avrà una velocità che varia in tutte le direzioni e in un ampio intervallo di valori. E’ quindi di interesse chiedersi quale sia la distribuzione statistica delle velocità. Questa distribuzione si può ottenere attraverso l’approccio della meccanica statistica, che abbiamo già accennato e di cui rivediamo adesso solo i principali presupposti..

40
Rivediamo i presupposti dell’approccio statistico
Prendiamo in considerazione un gas dal punto di vista microscopico. Si tratta di un sistema composto da N molecole in movimento. Con una certa astrazione, possiamo immaginare che le 3 coordinate spaziali di ogni molecola x, y, z e le 3 componenti della velocità di ogni molecola vx vy vz rappresentino un punto in uno spazio a 6 dimensioni. Chiameremo questo spazio a 6 dimensioni «spazio delle fasi». Supponiamo di dividere lo spazio delle fasi ( x , y , z , vx ,vy ,vz ) in tanti volumetti (celle) di dimensioni dx , dy , dz , dvx ,dvy ,dvz Il prodotto dx x dy x dz x dvx x dvy x dvz rappresenta il volume H di ogni cella In ogni cella i ci sarà un numero Ni di molecole, cioè di punti dello spazio delle fasi le cui coordinate sono all’interno della cella in questione.
 in tanti volumetti (celle) di dimensioni dx , dy , dz , dvx ,dvy ,dvz. Il prodotto dx x dy x dz x dvx x dvy x dvz rappresenta il volume H di ogni cella. In ogni cella i ci sarà un numero Ni di molecole, cioè di punti dello spazio delle fasi le cui coordinate sono all’interno della cella in questione.")
41
ρ = ρ (dx , dy , dz , dvx ,dvy ,dvz )
Potremo definire quindi una densità di punti nello spazio delle fasi data dalla: ρ = 𝑁𝑖 𝐻 Questa densità ρ sarà funzione delle 6 coordinate nello spazio delle fasi ρ = ρ (dx , dy , dz , dvx ,dvy ,dvz ) L’obiettivo della meccanica statistica (che studia proprio la termodinamica in termini microscopici) è quello di determinare questa funzione in termini probabilistici. Di questo approccio daremo solo alcuni cenni
 L’obiettivo della meccanica statistica (che studia proprio la termodinamica in. termini microscopici) è quello di determinare questa funzione in termini probabilistici. Di questo approccio daremo solo alcuni cenni.")
42
H dello spazio delle fasi costituisce un macro stato.
In questo approccio, e cioè uno spazio delle fasi diviso in un certo numero di cellette elementari di dimensioni H = dx x dy x dz x dvx x dvy x dvz , l’individuazione del numero di molecole presenti in ogni volumetto H ci fornisce a tutti gli effetti le proprietà del gas. Infatti, non è per niente rilevante quali molecole siano presenti in un dato volumetto, ma quante ce ne siano. Questo è facile da intuire: la pressione di un gas dipenderà da quante molecole hanno una certa velocità (e non quali), analogamente la densità di un gas dipenderà da quante molecole si trovano in un dato volume del gas (e non quali). La determinazione del numero Ni di molecole presenti ad ogni istante t in un volumetto H dello spazio delle fasi costituisce un macro stato. L’individuazione di quali molecole di trovino nel volumetto in questione costituisce invece un micro stato
, analogamente la densità di un gas dipenderà da quante. molecole si trovano in un dato volume del gas (e non quali). La determinazione del numero Ni di molecole presenti ad ogni istante t in un volumetto. H dello spazio delle fasi costituisce un macro stato. L’individuazione di quali molecole di trovino nel volumetto in questione costituisce. invece un micro stato.")
43
tutti i micro stati di un gas sono egualmente probabili
Il postulato della meccanica statistica applicata ai sistemi termodinamici è che tutti i micro stati di un gas sono egualmente probabili Questo a prima vista potrebbe sembrare curioso e tutto sommato non corretto: Consideriamo per esempio un micro stato in cui le molecole m1, m2, m3, …….mk si trovano distribuite come in figura: 1 3 5 7 9 11 2 13 4 6 8 10 12 14 15 17 19 21 23 25 27 16 18 20 22 24 26 28 29 31 33 35 37 39 30 41 32 34 36 38 40 42

44
è più probabile del micro stato seguente:
A prima vista saremmo indotti a pensare che il micro stato che abbiamo appena considerato è più probabile del micro stato seguente: 5 15 11 1 3 6 16 12 7 2 4 13 9 8 14 10 17 19 39 25 18 20 40 27 26 21 23 33 28 22 24 34 31 32 37 30 38 29 41 35 36 42 In realtà non è vero: la probabilità di occorrenza dei due micro stati è la stessa e vediamo perché

45
eguale al precedente, ma non lo è.
La probabilità di occorrenza del primo micro stato che abbiamo considerato è molto bassa 1 3 5 7 9 11 13 2 4 6 8 10 12 14 15 17 19 21 23 25 27 16 18 20 22 24 26 28 29 31 33 35 37 39 30 41 32 34 36 38 40 42 E non differisce dalla probabilità di occorrenza del seguente altro micro stato che sembra eguale al precedente, ma non lo è. 1 2 5 4 9 11 3 12 7 6 8 10 13 14 15 17 27 21 23 25 19 16 18 20 22 24 26 28 41 31 37 35 33 39 30 29 32 34 36 38 40 42

46
2 particelle per ogni cella
La probabilità di occorrenza di ognuno di questi tre micro stati è molto bassa ma è eguale per tutti e tre in quanto per tutti e tre stiamo specificando le coordinate di ogni singola particella. Non c’è dubbio però che i primi due micro stati hanno qualcosa in comune: 2 particelle per ogni cella Cioè: i primi due micro stati corrispondono allo stesso macro stato 1 3 5 7 9 11 13 2 4 6 8 10 12 14 15 17 19 21 23 25 27 16 18 20 22 24 26 28 29 31 33 35 37 39 30 41 32 34 36 38 40 42 1 2 5 4 9 11 3 12 7 6 8 10 13 14 15 17 27 21 23 25 19 16 18 20 22 24 26 28 41 31 37 35 33 39 30 29 32 34 36 38 40 42 5 15 11 1 3 6 16 12 7 13 2 4 9 8 14 10 17 19 39 25 18 20 40 27 26 21 23 33 28 22 24 34 31 32 37 30 38 29 41 35 36 42

47
In un gas in equilibrio, ci sono un numero elevatissimo di micro stati che corrispondono
allo stesso macro stato e sono egualmente probabili e il sistema passa continuamente da un micro stato all’altro senza alterare le variabili macroscopiche del sistema.

48
N = numero totale di molecole m = massa di ogni singola molecola
Una interessante distribuzione statistica, basata proprio su questi presupposti, è stata ricavata da Maxwell che ha dimostrato che: Dove: N v dv = numero di molecole che hanno una velocità compresa fra v e v+dv N = numero totale di molecole m = massa di ogni singola molecola k = costante di Boltzman T = temperatura

49
Tipica forma della curva maxwelliana per due diversi valori di temperatura:
Il numero di molecole aventi velocità fra v e v + Δv sarà l’area sottesa dalla corrispondente curva fra gli intervalli di velocità in questione.

50
In questo grafico notiamo che a temperatura più elevata la velocità più probabile, vp,
aumenta, in accordo col significato microscopico di temperatura, e cioè di «agitazione termica». Va comunque notato che aumenta anche la dispersione dei valori di velocità, cioè aumenta in percentuale il numero di molecole aventi v velocità superiori al valore più probabile.

51
Conferma sperimentale della distribuzione di Maxwell
Esperimento di Stern (1926)
")
52
Risultati dell’esperimento di Stern

Presentazioni simili



















:>")