Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

3
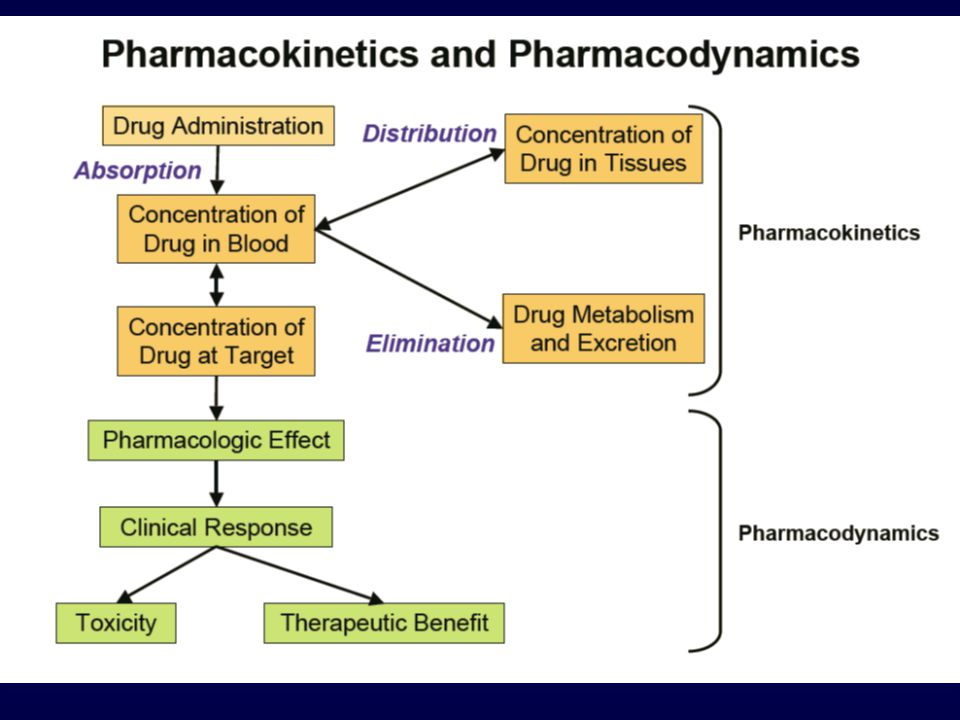
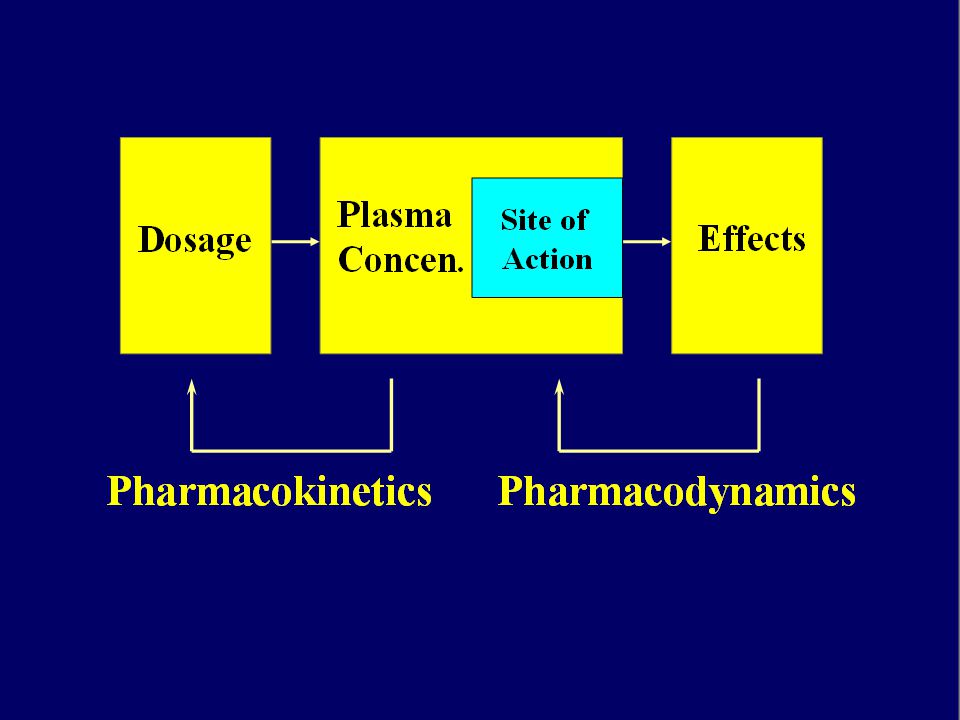
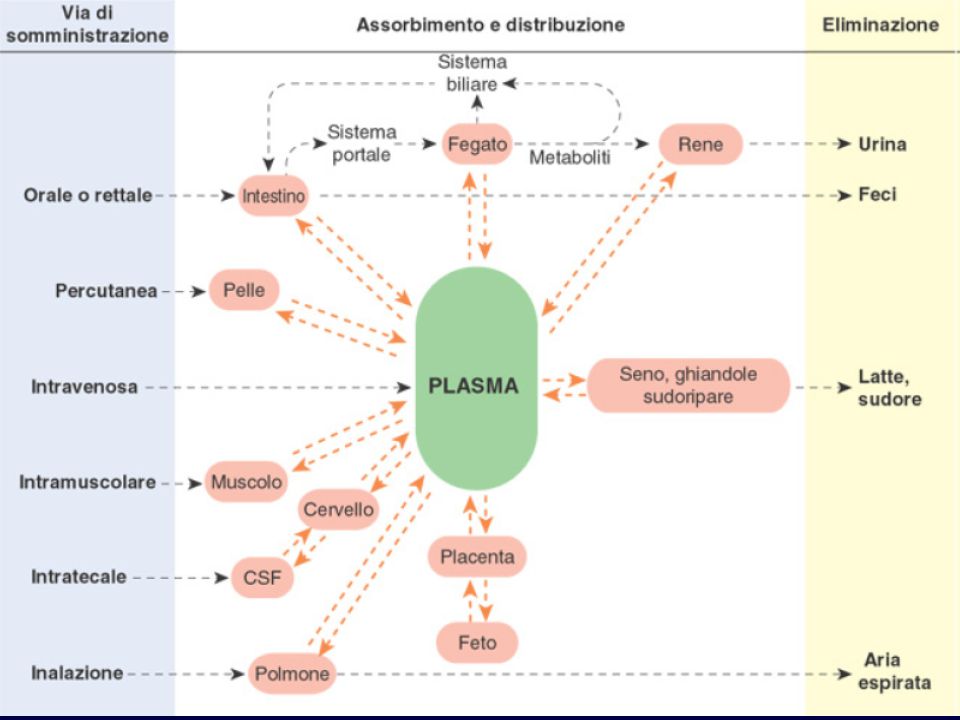
FARMACOCINETICA Studia l’evoluzione temporale delle concentrazioni di un farmaco e dei suoi metaboliti nei diversi fluidi e tessuti dell’organismo mediante l’analisi dei processi che ne regolano: ASSORBIMENTO DISTRIBUZIONE METABOLISMO ELIMINAZIONE ADME

4
“ciò che l’uomo fa al farmaco”
FARMACOCINETICA Relazioni matematiche che permettono di stabilire le dosi e la posologia mediante l’integrazione dei concetti di: assorbimento/distribuzione/metabolismo/escrezione “ciò che l’uomo fa al farmaco” - viene valutata su studi preclinici e su studi di fase I -

5
La FARMACOCINETICA CLINICA permette al
medico di aggiustare la posologia, stabilita negli individui sani, in funzione delle modificazioni fisiologiche e patologiche che si verificano negli individui ammalati

6
INDIVIDUALIZZATA E RAZIONALE
Scopo finale... UNA TERAPIA INDIVIDUALIZZATA E RAZIONALE

7
OBIETTIVI: Sviluppare nuovi farmaci
Selezionare la via di somministrazione Scegliere la forma farmaceutica Conoscere la capacità di accesso ad organi e tessuti Conoscere le vie metaboliche Caratterizzare i processi di eliminazione Stabilire le relazioni con la risposta farmacologica Migliorare i risultati dei trattamenti

8
DOSE ORALE DOSE I.V. TRATTO G.I. CIRCOLAZIONE SISTEMICA DISTRIBUZIONE PERIFERICA FEGATO ORGANO BERSAGLIO CLEARANCE RECETTORE EFFETTO

9
The disposition of chemicals entering the body (from C. D
The disposition of chemicals entering the body (from C.D. Klaassen, Casarett and Doull’s Toxicology, 5th ed., New York: McGraw-Hill, 1996).
.")
10
Passaggio del farmaco dalla sede si somministrazione al sangue
ASSORBIMENTO Passaggio del farmaco dalla sede si somministrazione al sangue

12
MEMBRANA CELLULARE Proteine periferiche Proteina integrale La membrana cellulare è costituita da un doppio strato fosfolipidico le cui teste idrofile formano le superfici interna ed esterna e le code idrofobe si uniscono al centro della membrana. Il doppio strato ha uno spessore di circa 4,5 nanometri. Le proteine, che costituiscono gli altri componenti della membrana, possono essere di due tipi. Alcune dette periferiche sono disposte su entrambe le facce della membrana, altre dette integrali penetrano nella membrana e l’attraversano completamente.

13
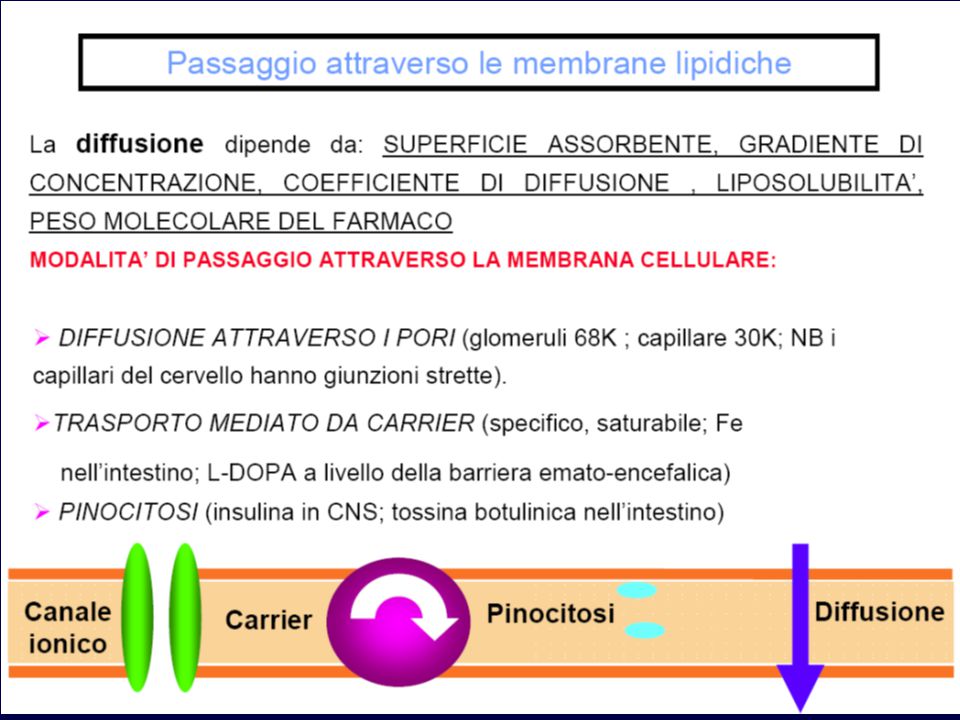
Movimento attraverso le membrane cellulari
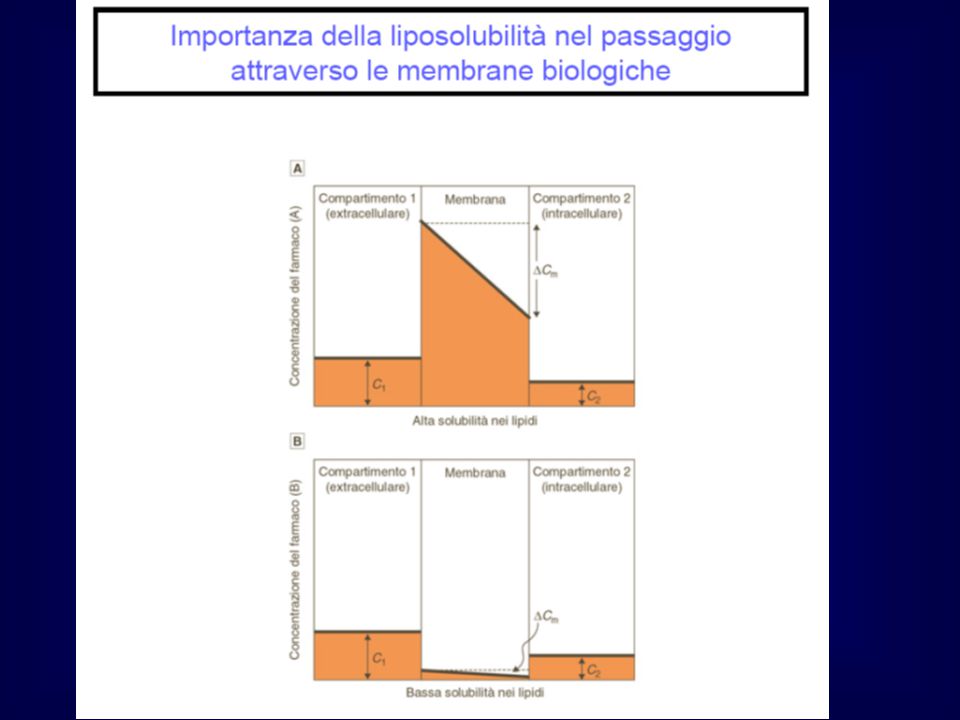
Il principale fattore che determina il grado di trasferimento per diffusione passiva attraverso le membrane è la solubilità dei farmaci nei lipidi. Il peso molecolare è un fattore meno importante Sostanze non polari sono altamente solubili nei solventi non polari come i lipidi Molti farmaci sono acidi o basi deboli; il loro grado di ionizzazione varia in funzione del pH (Henderson Hasselbalch) Solo la forma non ionizzata può diffondere attraverso le membrane lipidiche; la sua concentrazione nei due compartimenti sarà influenzata dalla differenza di pH
 Solo la forma non ionizzata può diffondere attraverso le membrane lipidiche; la sua concentrazione nei due compartimenti sarà influenzata dalla differenza di pH.")
17
Proteine ed altre grosse molecole
Passaggio dei farmaci attraverso le membrane biologiche in funzione delle loro caratteristiche chimico-fisiche Caratteristiche del farmaco Passaggio attraverso le membrane biologiche PROCESSO PASSIVO Sostanze idrosolubili, non ionizzabili, con diametro molecolare inferiore a 4 Å (acqua, urea, alcool) - Filtrazione attraverso i pori Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata. Il trasferimento dipende dal pKa della sostanza e dal gradiente di pH ai due lati della membrana MECCANISMO DI TRASPORTO Sostanze idrosolubili non ionizzate con diametro superiore a 4 Å (glucosio) - Diffusione facilitata senza dispendio energetico per mezzo di un trasportatore Acidi e basi organiche ionizzate -Trasporto attivo con dispendio energetico mediante un trasportatore Proteine ed altre grosse molecole - Fagocitosi e pinocitosi (trasporto vescicolare)
 - Filtrazione attraverso i pori. Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata. Il trasferimento dipende dal pKa della sostanza e dal gradiente di pH ai due lati della membrana. MECCANISMO DI TRASPORTO. Sostanze idrosolubili non ionizzate con diametro superiore a 4 Å (glucosio) - Diffusione facilitata senza dispendio energetico per mezzo di un trasportatore. Acidi e basi organiche ionizzate. -Trasporto attivo con dispendio energetico mediante un trasportatore. Proteine ed altre grosse molecole. - Fagocitosi e pinocitosi (trasporto vescicolare)")
19
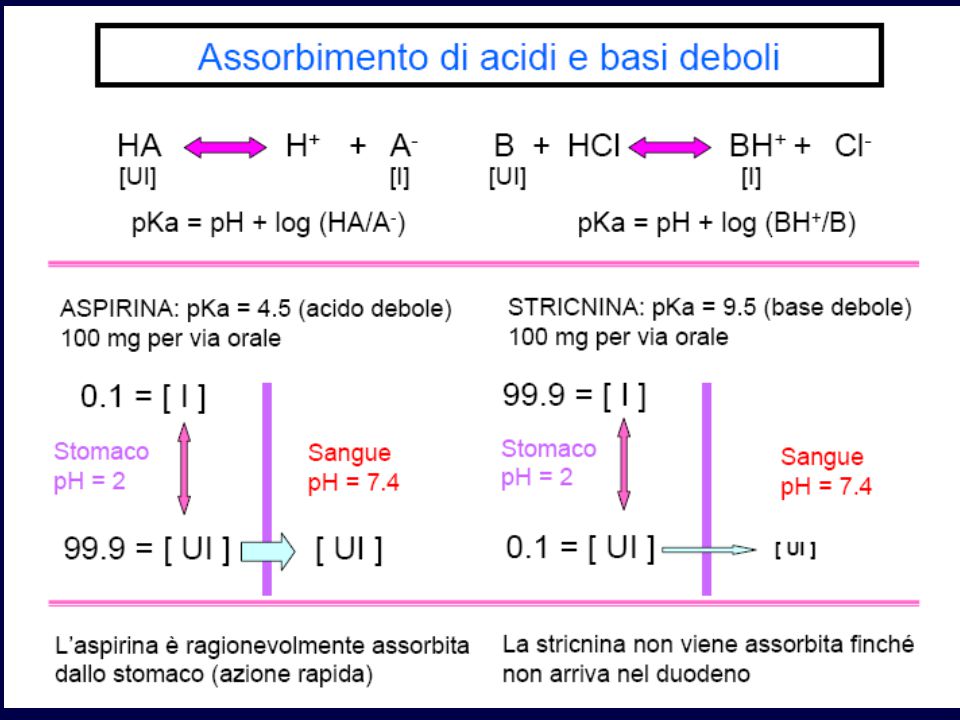
ASSORBIMENTO GASTRICO DI UN SOSTANZA ACIDA
(pKa = 3,4) Stomaco: pH = 1,4 Plasma: pH = 7,4
 Stomaco: pH = 1,4. Plasma: pH = 7,4.")
24
Intrappolamento ionico - aspirina
Aspirina è un acido debole con pKa = 3.5 Nello stomaco (pH 2-3), la maggior parte del farmaco è non ionizzata Nell’intestino (pH 5-6), più ionizzata aspirina assorbita meglio nello stomaco Nel sangue (pH 7.4), quasi tutta ionizzata una volta che l’aspirina va dallo stomaco al sangue è “intrappolata” nel sangue (non torna facilmante nello stomaco)
, la maggior parte del farmaco è non ionizzata. Nell’intestino (pH 5-6), più ionizzata. aspirina assorbita meglio nello stomaco. Nel sangue (pH 7.4), quasi tutta ionizzata. una volta che l’aspirina va dallo stomaco al sangue è intrappolata nel sangue (non torna facilmante nello stomaco)")
26
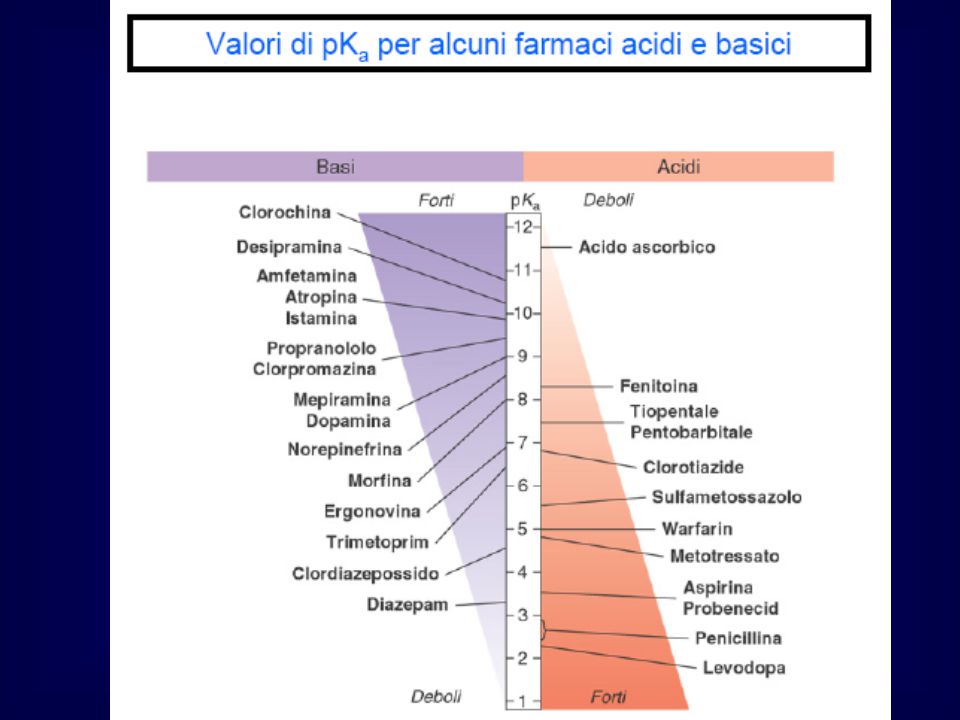
FATTORI CHE CONDIZIONANO L’ASSORBIMENTO
DI UN FARMACO Caratteristiche del farmaco: massa molecolare, stato fisico, carica, stabilità, solubilità…. Proprietà dell’organismo: morfologia e dimensioni della superficie assorbente, perfusione dell’area assorbente, specie, razza, età, stato nutrizionale, stato di salute….. Caratteristiche dell’esposizione: dose, via di somministrazione, durata del contatto con la superficie assorbente…. Fattori esogeni: formulazione, interazione con altre sostanze, condizioni fisiche (es. temperatura)…..
…..")
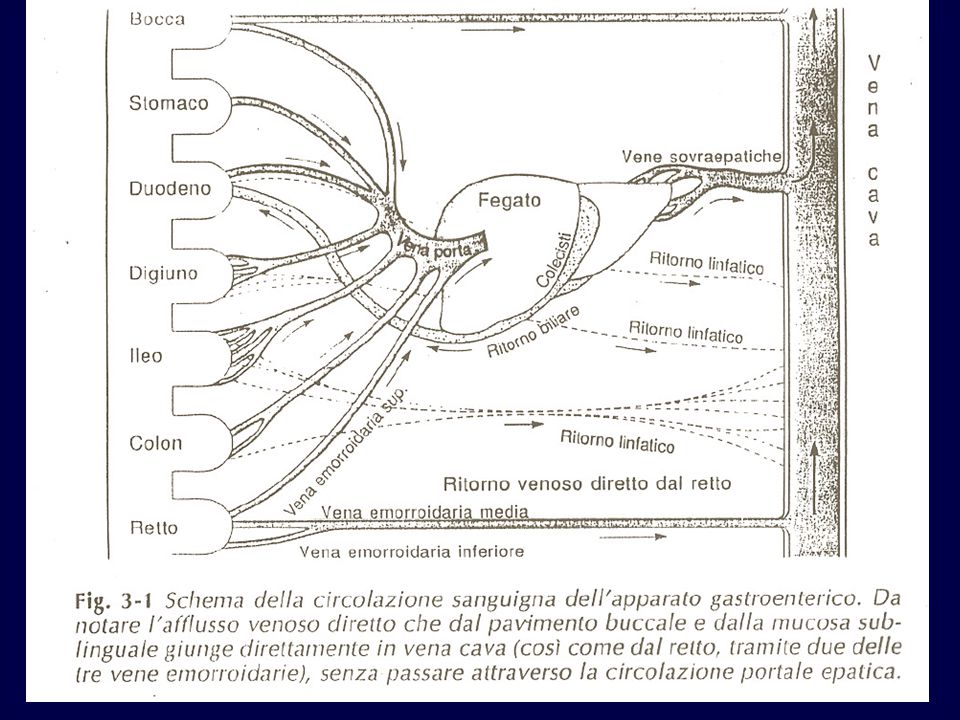
27
I capillari sanguiferi hanno un’organizzazione morfo-funzionale diversa a seconda della sede in cui si trovano QUINDI: la permeabilità del letto vascolare ad un certo farmaco è diversa a seconda del distretto irrorato

28
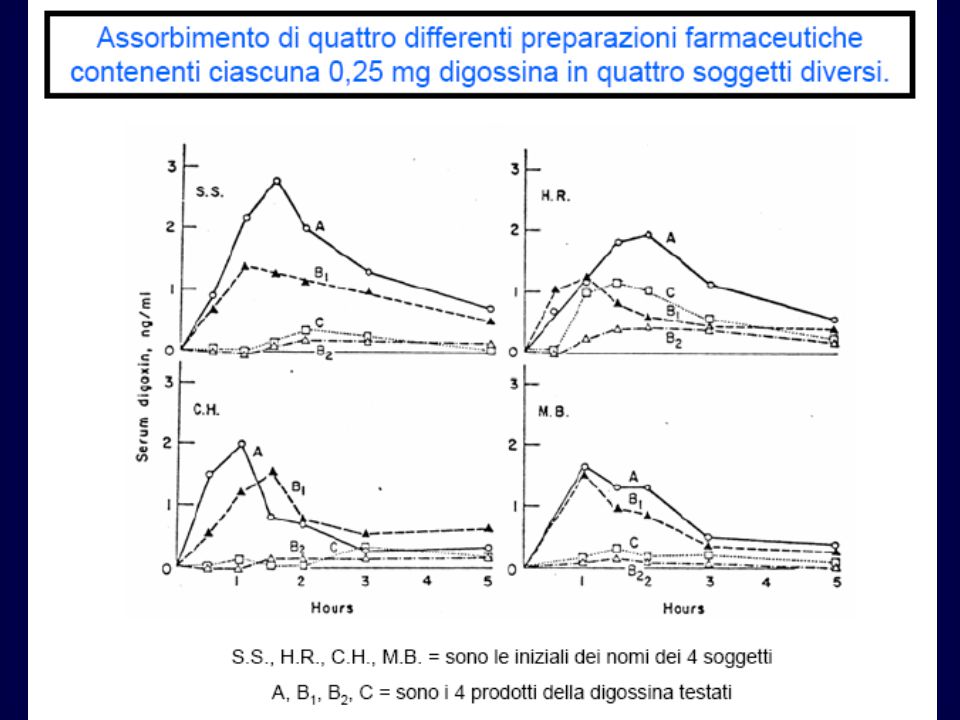
Biodisponibilità Rappresenta la percentuale di farmaco che
è reso disponibile all’organismo L’iniezione intravenosa del farmaco determina una biodisponibilità del 100% La biodisponibilità per le altre preparazioni farmaceutiche viene calcolata confrontando l’area sotto la curva (AUC) relativa alla concentrazione plasmatica del farmaco somministrato endovena o attraverso altre vie
 relativa alla concentrazione plasmatica del farmaco somministrato endovena o attraverso altre vie.")
29
Distrutta nell’intestino Distrutta dalla parete intestinale
Biodisponibilità Distrutta nell’intestino Non assorbita Distrutta dalla parete intestinale Distrutta dal fegato Circolazione sistemica Dose

32
VIE DI SOMMINISTRAZIONE
ORALE CUTANEA POLMONARE RETTALE MAMMARIA CONGIUNTIVALE NATURALI ENDOVENOSA INTRAMUSCOLARE SOTTOCUTANEA INTRAPERITONEALE EPIDURALE INTRARTICOLARE INTRAMIDOLLARE INTRARTERIOSA ARTIFICIALI Vie parenterali: al di fuori del tratto gastroenterico

34
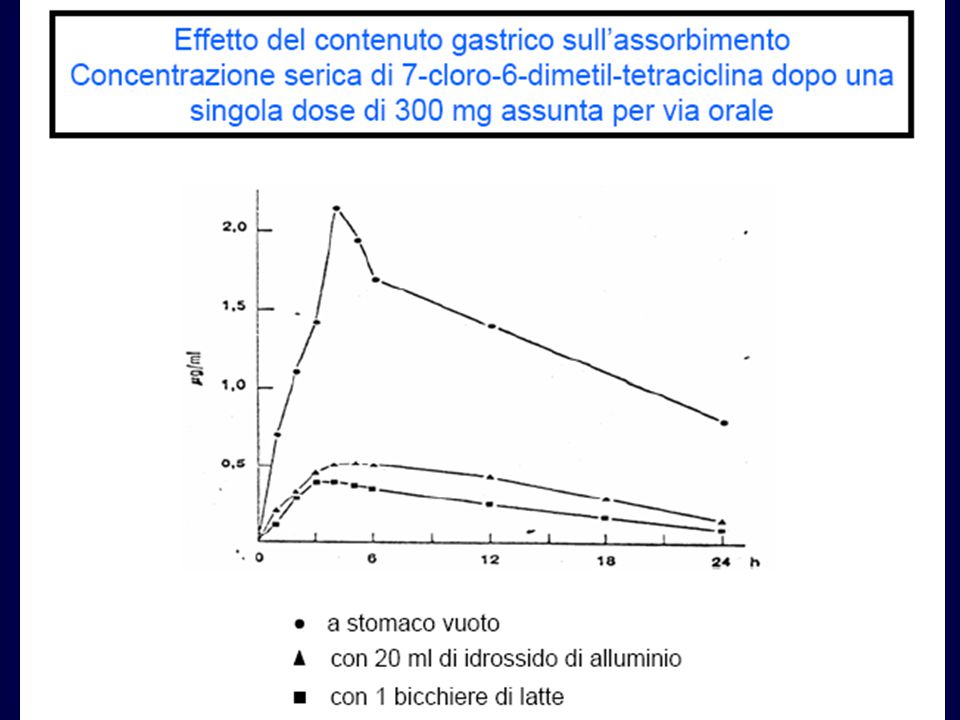
Fattori che influenzano l’assorbimento orale
Disintegrazione della preparazione farmaceutica Dissoluzione delle particelle Stabilità chimica del farmaco Stabilità del farmaco alla degradazione enzimatica Motilità e mescolamento nel tratto GI Presenza e tipo di cibo Passaggio attraverso la parete GI Flusso sanguigno nel tratto GI Tempo di svuotamento dello stomaco FORMULAZIONE

35
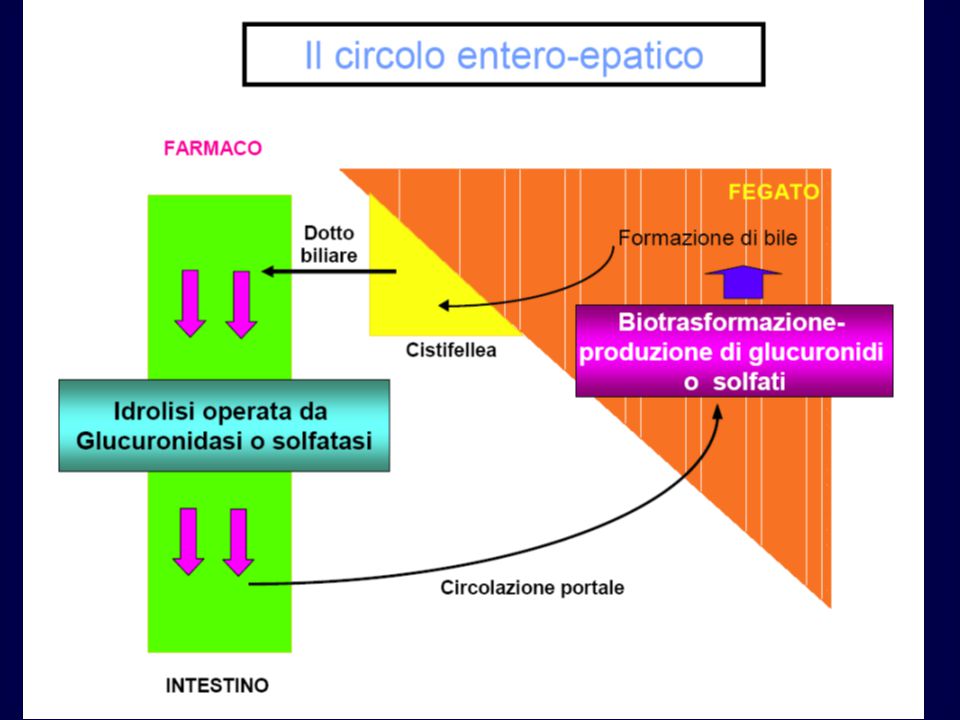
FATTORI CHE CONDIZIONANO L’ASSORBIMENTO
GASTROINTESTINALE Legge di azione di massa Equazione di Henderson-Hasselbach Fase farmaceutica (disintegrazione e dissoluzione) Area della superficie di assorbimento Velocità del flusso ematico Resistenza al pH gastrico, agli enzimi dello stomaco, dell’intestino e della flora intestinale Trasporto specializzato Circolo enteroepatico Effetto di primo passaggio
 Area della superficie di assorbimento. Velocità del flusso ematico. Resistenza al pH gastrico, agli enzimi dello stomaco, dell’intestino. e della flora intestinale. Trasporto specializzato. Circolo enteroepatico. Effetto di primo passaggio.")
41
ASSORBIMENTO POLMONARE (gas, vapori e liquidi volatili)
Cellule epiteliali degli alveoli ed endotelio dei capillari ampiamente fenestrati Flusso ematico elevato Coefficiente di ripartizione liquido/gas Rapporto di solubilità sangue/gas (etilene) Assorbimento con maggiore velocità di circolo ematico Rapporto di solubilità sangue/gas (cloroformio) Assorbimento con maggiore frequenza e profondità del respiro
 Assorbimento con maggiore velocità di circolo ematico. Rapporto di solubilità sangue/gas (cloroformio) Assorbimento con maggiore frequenza e profondità del respiro.")
45
Preparazioni a rilascio prolungato
Iniezioni deposito (oleose, viscose o in particelle) Compresse multisrato (gastroresistenti) Capsule a rilascio prolungato Infusori (con o senza sensori) Cerotti (nicotina, GTN) Pro-farmaci Liposomi Farmaci con bersaglio o diretti contro anticorpi
 Compresse multisrato (gastroresistenti) Capsule a rilascio prolungato. Infusori (con o senza sensori) Cerotti (nicotina, GTN) Pro-farmaci. Liposomi. Farmaci con bersaglio o diretti contro anticorpi.")
47
CINETICHE DI ASSORBIMENTO
CINETICA DI I ORDINE: LA QUANTITA’ DI FARMACO ASSORBITA NELL’UNITA’ DI TEMPO E’ UNA % COSTANTE DI QUELLA CHE RIMANE DA ASSORBIRE La maggior parte dei farmaci diffonde per diffusione passiva, seguendo una cinetica di I ordine CINETICA DI ORDINE 0: LA QUANTITA’ DI FARMACO ASSORBITA NELL’UNITA’ DI TEMPO E’ COSTANTE Per alcuni farmaci l’assorbimento avviene attraverso meccanismi di trasporto attivo saturabili (la velocità di assorbimento dipende dal numero di trasportatori), seguendo una cinetica di ordine 0
, seguendo una cinetica di ordine 0.")
48
CINETICHE DI ASSORBIMENTO
La cinetica di I ordine indica che la quantità di farmaco assorbita in ogni istante è proporzionale alla quantità di farmaco che resta da assorbire; se invece una quantità costante viene assorbita nell’unità di tempo, come avviene per un processo di trasporto attivo saturato, la cinetica viene detta cinetica di ordine 0. Le cinetiche di I ordine sono molto più frequenti nei procesi dominati dalla diffusione e da altri fenomeni passivi, sono cioè molto più naturali e frequenti rispetto alle cinetiche di ordine 0, che richiedono la presenza di processi di trasporto attivi. Un farmaco assorbito per trasporto attivo mostra una cinetica di eliminazione di ordine 0 solo se il trasporto attivo è saturato e lavora a pieno regime, mentre se è sotto saturato, il trasporto tende a divenire proporzionale al legame tra farmaco e trasportatore e perciò alla concentrazione del farmaco, cioè segue una cinetica di I ordine.

49
CINETICHE DI I ORDINE Se un farmaco ha una concentrazione di 8 g/l e ne viene assorbito 1/10 nell’unità di tempo (minuto), dopo un minuto ne rimarrà da assorbire = 7,2 g/l e così via. Ragionando in termini di frazione, se 8 gr/l è = 1, dopo 1 minuto resterà 0,9, dopo 2 minuti 0,9 - 0,09 = 0,81, ecc. In generale, se una frazione k viene assorbita nell’unità di tempo, ne resterà una frazione (1-k)t dopo t minuti. Trasformando la curva in scala semilogaritmica, possiamo introdurre un numero, chiamato e in modo tale che se k è un numero molto piccolo (k<<1), 1-k = e-k, così la frazione che resta da assorbire al tempo t sarà e-kt. Il log in base e (log) di questa frazione è -kt. 8 4 2 1 1 0.9 0.8 0.5 0.25 0.125 Concentrazione (gr/l) Concentrazione/conc. iniziale , , ,7 tempo (minuti)
, dopo un minuto ne rimarrà da assorbire = 7,2 g/l e così via. Ragionando in termini di frazione, se 8 gr/l è = 1, dopo 1 minuto resterà 0,9, dopo 2 minuti 0,9 - 0,09 = 0,81, ecc. In generale, se una frazione k viene assorbita nell’unità di tempo, ne resterà una frazione (1-k)t dopo t minuti. Trasformando la curva in scala semilogaritmica, possiamo introdurre un numero, chiamato e in modo tale che se k è un numero molto piccolo (k<<1), 1-k = e-k, così la frazione che resta da assorbire al tempo t sarà e-kt. Il log in base e (log) di questa frazione è -kt Concentrazione (gr/l) Concentrazione/conc. iniziale ,9 13,8 20,7. tempo (minuti)")
50
CINETICHE DI I ORDINE Se trasformiamo il grafico precedente in scala logaritmica, il numero e diventa la base dei logaritmi naturali. Se mettiamo in grafico il logaritmo della concentrazione iniziale (C0) rispetto al tempo, l’equazione sarà: log (C0 x e-kt) = log C0 - kt cioè una retta che partendo dal logaritmo di C0 scende con pendenza k rispetto al tempo. k è definito anche come costante di assorbimento, cioè la frazione assorbita nell’unità di tempo. La cinetica può essere descritta come: C = C0 x e-kt 8 4 2 1/e 2,079 1,386 0.693 -1 Log (concentrazione) Concentrazione (gr/l) , , , ,7 tempo (minuti)
 rispetto al tempo, l’equazione sarà: log (C0 x e-kt) = log C0 - kt. cioè una retta che partendo dal logaritmo di C0 scende con pendenza k rispetto al tempo. k è definito anche come costante di assorbimento, cioè la frazione assorbita nell’unità di tempo. La cinetica può essere descritta come: C = C0 x e-kt /e. 2,079. 1, Log (concentrazione) Concentrazione (gr/l) 0 6,93 13,8 20,9 27,7. tempo (minuti)")
51
C = C0 x e-kt C/C0 = e-kt 1/2 = e-kt
CINETICHE DI I ORDINE k è definito anche come costante di assorbimento, cioè la frazione assorbita nell’unità di tempo. Il suo inverso 1/k, definito t è detta costante di tempo dell’assorbimento. Un utile parametro per definire l’andamento della concentrazione del farmaco è il tempo necessario per dimezzarne la concentrazione nel sito di assorbimento, definito come TEMPO DI DIMEZZAMENTO, ovvero EMIVITA di assorbimento, cioè quando C/C0 = 1/2 QUINDI: C = C0 x e-kt C/C0 = e-kt /2 = e-kt t1/2 = log (2)/k t1/2 = log(2) x 1/k t1/2 = t x 0,693
/k t1/2 = log(2) x 1/k t1/2 = t x 0,693.")
52
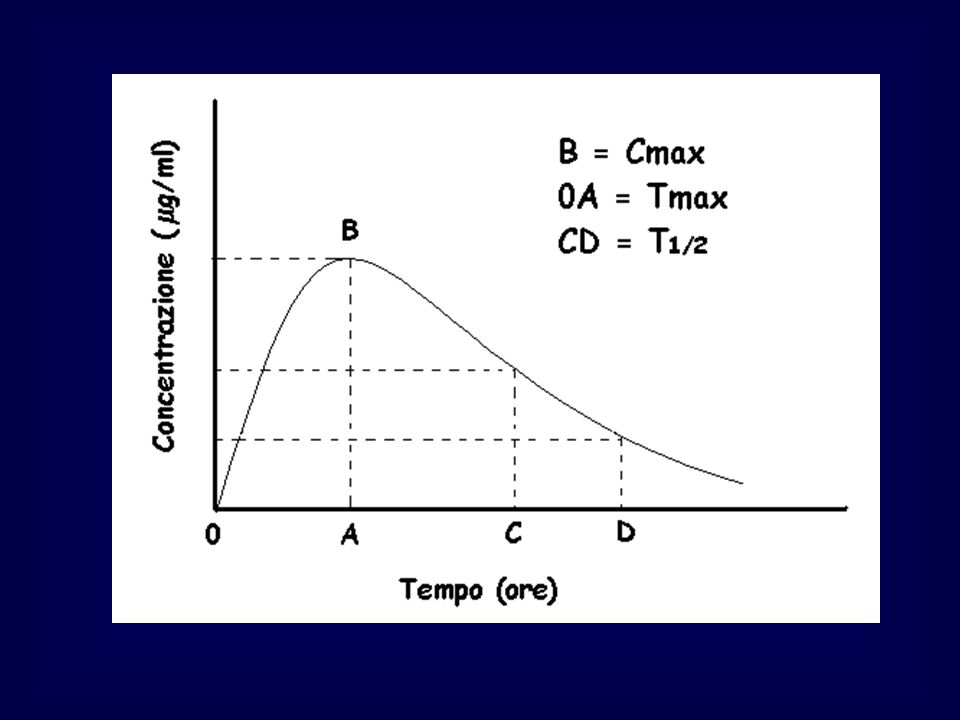
La velocità di assorbimento determina il picco plasmatico ed il tempo necessario per raggiungerlo
Un farmaco viene somministrato in due muscoli con irrorazione diversa. Nel primo caso (grafici A e C) l’assorbimento ha emivita di 28’, nel secondo (grafici B e D) di 84’. Le diverse velocità di assorbimento (linea tratteggiata) influenzano sia i flussi di eliminazione (linea continua) che le concentrazioni plasmatiche (C e D). Il tempo di picco corrisponde al momento in cui i flussi di assorbimento e di eliminazione hanno pari valore.
 l’assorbimento ha emivita di 28’, nel secondo (grafici B e D) di 84’. Le diverse velocità di assorbimento (linea tratteggiata) influenzano sia i flussi di eliminazione (linea continua) che le concentrazioni plasmatiche (C e D). Il tempo di picco corrisponde al momento in cui i flussi di assorbimento e di eliminazione hanno pari valore.")
53
La velocità di assorbimento varia a seconda della via di somministrazione utilizzata
La concentrazione plasmatica di un farmaco nell’unità di tempo dipende dalla differenza tra la quantità assorbita e la quantità eliminata Il picco di concentrazione plasmatica di un farmaco dipende dalla velocità di assorbimento: più lento è l’assorbimento, più basso è il picco plasmatico

54
Route for administration
-Time until effect- intravenous seconds intraosseous seconds endotracheal 2-3 minutes inhalation 2-3 minutes sublingual 3-5 minutes intramuscular minutes subcutaneous minutes rectal 5-30 minutes ingestion minutes transdermal (topical) variable (minutes to hours)
 variable (minutes to hours)")
Presentazioni simili






















, specializzato per la distribuzione di: gas.>")








