Scaricare la presentazione
1
A.A CORSO BIOINFORMATICA 2 LM in BIOLOGIA EVOLUZIONISTICA Scuola di Scienze, Università di Padova Docenti: Dr. Giorgio Valle Dr. Stefania Bortoluzzi

2
DEFINIZIONI DI BIOINFORMATICA
The application of computer technology to organize and analyze biological data. Analysis of proteins, genes, and genomes using computer algorithms. NIH: “research, development, or application of computational tools and approaches for expanding the use of biological, medical, behavioral or health data, including those to acquire, store, analyze, or visualize such data”

3
Computational Biology
BIOINFORMATICA Bioinformatics Medical Informatics Computational Biology Genomics Proteomics Pharmacogenomics Evolutionary Biology

4
BIOINFORMATICA The study and application of computing methods for classical biology SINONIMO BIOINFORMATICS A SUA VOLTA MOLTE DISCIPLINE FIGLIE

5
BIOINFORMATICA Computational and Statistical Genetics. Analysis and comparison of the entire genome of a single species or of multiple species

6
BIOINFORMATICA Study of how the genome is expressed in proteins, and of how these proteins function and interact

7
BIOINFORMATICA Pharmacogenomics is the study of how genes affect a person’s response to drugs. This relatively new field combines pharmacology (the science of drugs) and genomics (the study of genes and their functions) to develop effective, safe medications and doses that will be tailored to a person’s genetic makeup. The application of genomic methods to identify drug targets, for example, searching entire genomes for potential drug receptors
 and genomics (the study of genes and their functions) to develop effective, safe medications and doses that will be tailored to a person’s genetic makeup. The application of genomic methods to identify drug targets, for example, searching entire genomes for potential drug receptors.")
8
BIOINFORMATICA Study of the evolutionary processes that produced the diversity of life, the descent of species, and the origin of new species.

9
BIOINFORMATICA The study and application of computing methods to improve communication, understanding, and management of medical data Biomedical informatics (BMI) is the interdisciplinary, scientific field that studies and pursues the effective uses of biomedical data, information, and knowledge for scientific inquiry, problem solving and decision making, motivated by efforts to improve human health.
 is the interdisciplinary, scientific field that studies and pursues the effective uses of biomedical data, information, and knowledge for scientific inquiry, problem solving and decision making, motivated by efforts to improve human health.")
10
BIOINFORMATICA: AMBITI PRINCIPALI ?
Sviluppo e implementazione di software per conservare, diffondere e elaborare diversi tipi di informazione Sviluppo di nuovi algoritmi e metodi statistici per integrare dati diversi, ricercare e studiare diversi tipi di relazioni e interazioni in grandi dataset Analisi e interpretazione di dati di varia natura, quali biosequenze, strutture, interazioni.

11
BIOINFORMATICA: SCOPI?
Aumentare la comprensione della biologia a livello funzionale Modellizzare il funzionamento delle cellule e degli organismi Fornire informazioni utili a migliorare la qualità della vita (malattie, tumori) Database (design, handling, ...) Analisi di dati d’espressione Predizione di funzioni geniche Identificazione di fattori di rischio per malattie Studio delle reti regolative e metaboliche … Mappaggio di geni e genomi Predizione di strutture Identificazione di target Drug design Terapia genica
 Database (design, handling, ...) Analisi di dati d’espressione. Predizione di funzioni geniche. Identificazione di fattori di rischio per malattie. Studio delle reti regolative e metaboliche. … Mappaggio di geni e genomi. Predizione di strutture. Identificazione di target. Drug design. Terapia genica.")
12
BIOINFORMATICA: QUALI DATI?
Sequence data Structural information Expression data Molecular interaction data Mutation data Phenotypic data Imaging data

13
BIOINFORMATICA 3 PROSPETTIVE
CELLULA ORGANISMO ALBERO DELLA VITA

14
I : CELLULA

15
I : CELLULA CENTRAL DOGMA OF MOLECULAR BIOLOGY
DNA RNA Proteina Trascrizione Traduzione Genoma Trascrittoma Proteoma CENTRAL DOGMA OF BIOINFORMATICS AND GENOMICS 15

16
I : CELLULA

17
Il ruolo della bioinfomatica
I : CELLULA Il ruolo della bioinfomatica Questi miliardi di sequenze presentano sfide e opportunità, per studiare moltissimi problemi biologici diversi, quali: Stati cellulari in relazione a ciclo cellulare, differenziamento, malattia ecc. Regolazione dei processi, …

18
II : ORGANISMO Tempo Sviluppo Spazio e stato
Regione del corpo, fisiologia, patologia, farmacologia

19
Il ruolo della bioinfomatica
II : ORGANISMO Il ruolo della bioinfomatica Studia il genoma, e particolarmente l’insieme di geni espressi in trascritti RNA e in prodotti proteici. Gli strumenti bioinformatici posso esser usati per descrivere cambiamenti qualitativi e qunatitativi degli elementi considerati: durante lo sviluppo, in relazione alle diverse zone del corpo, e in una serie molto vasta di stati fisiologici o patologici.

20
III : ALBERO DELLA VITA Darwin (1837) Haeckel (1866)
l’idea di evoluzione nella concettualizzazione dei fenomeni biologici e la sua progressiva ridefinizione sulla base degli sviluppi teorici e tecnici delle scienze biologiche sono tra le più significative conquiste intellettuali della scienza “The green and budding twigs may represent existing species; and those produced during former years may represent the long succession of extinct species.” «in biologia niente ha senso se non alla luce dell’evoluzione» (Dobzhansky, 1973)
")
21
TREE OF LIFE WITH ENDOSYMBIOSIS
III : ALBERO DELLA VITA Gli esseri viventi oggi esistenti si sono evoluti a partire da altri esseri viventi ancestrali e sono legati da relazioni di tipo evolutivo Lo studio del patrimonio genetico (genoma) delle specie permette di ricostruirne la storia passata e le relazioni con altre specie alberi filogenetici TREE OF LIFE WITH ENDOSYMBIOSIS
 delle specie permette di ricostruirne la storia passata e le relazioni con altre specie alberi filogenetici. TREE OF LIFE WITH ENDOSYMBIOSIS.")
22
Il ruolo della bioinfomatica
III : ALBERO DELLA VITA Il ruolo della bioinfomatica Storicamente l’evoluzione molecolare è stato il primo ambito che ha richiesto al nascita della bioinformatica Studiare i processi evolutivi (da macro- a micro-evoluzione) Ricostruire la storia passata Comprendere le pressioni evolutive in relazione alle informazioni funzionali e viceversa
 Ricostruire la storia passata. Comprendere le pressioni evolutive in relazione alle informazioni funzionali e viceversa.")
23
DATABASES AND DATA RETRIEVAL Biosequences and Gene-related info

24
Alfabeto molecolare GLI ACIDI NUCLEICI E LE PROTEINE SONO POLIMERI LINEARI BIOSEQUENZE DNA e RNA sono polimeri lineari di nucleotidi, specializzati nel deposito, nella trasmissione e nell’utilizzazione dell’informazione genetica Gli acidi nucleici possono assumere specifiche forme nello spazio 3D, come le proteine, e svolgere attivita’ diverse (ad es. catalisi) IL DOGMA CENTRALE DELLA BIOLOGIA Molti altri RNA non coding
 IL DOGMA CENTRALE DELLA BIOLOGIA. Molti altri RNA non coding.")
25
THE BIG DATA ERA La biologia molecolare è nell'era dei "big data”
Le metodologie sperimentali high-troughput permettono di studiare moltissimi processi su scala genomica La grande disponibilità di dati sperimentali e conoscenza richiedono approcci quantitativi basati sull'informatica e la statistica per lo studio dei fenomeni biologici.

26
Evoluzione delle tecnologie di sequenziamento
Deep sequencing Evoluzione delle tecnologie di sequenziamento Standard Sanger 1° generation: Next Generation Sequencing (NGS) 2° generation: Ion Torrent, Nanopore 3° generation: Se oltre 7 minuti saltare quella dopo Roche/454 Illumina/Solexa Oxford Nanopore ABI/Solid LifeTechnologies’ IonTorrent Since: ‘70s Read length: bp bp bp Throughput: 300kb/run Mb/day Gb/day
 2° generation: Ion Torrent, Nanopore. 3° generation: Se oltre 7 minuti saltare quella dopo. Roche/454. Illumina/Solexa. Oxford Nanopore. ABI/Solid. LifeTechnologies’ IonTorrent. Since: ‘70s Read length: 1000 bp bp 100 bp. Throughput: 300kb/run Mb/day 12Gb/day.")
27
Millions of sequences (raw reads) per sample
Cominciamo con uno scorcio sull’evoluzione delle tecnologie di sequenziamento. Fin dagli anni settanta (‘75 o ‘77) è stato utilizzato il metodo Sanger. (chain termination method, dideoxynucleotides ddNTPs) Questa tecnica nel corso degli anni è stata perfezionata ed automatizzata fino a raggiungere sequenze, o reads, lunghe fino a mille paia di basi, e fino ad un totale di trecento (384) sequenze per esperimento. Il metodo Sanger richiede però lunghi tempi di preparazione dei campioni (clonazione) e la quantità di paia di basi sequenziate è relativamente bassa. A partire dal 2004 sono state introdotte le tecnologie di sequenziamento chiamate Next Generation Sequencing, o NGS, sviluppate da compagnie come Roche/454 , Applied Biosystems/Solid o Illumina/Solexa. La caratteristiche principali di queste tecnologie sono la brevità delle reads sequenziate, in un range tra 35 e 1000 paia di basi a seconda della macchina utilizzata, e l’alto parallelismo automatizzato dei processi di sequenziamento, che permette di ottenere un’enorme quantità di basi sequenziate per ogni esperimento. Per questa caratteristica ci si riferisce al Next generation Sequencing anche con il termine di Massive Parallel Sequencing. Confrontando con il metodo della prima generazione vediamo come il numero di paia di basi sequenziate è da uno a due ordini di grandezza superiore. Già dallo scorso anno sono state presentate nuove tecnologie che rappresentano un’ulteriore evoluzione dei metodi NGS, come l’Ion Torrent della Life Technologies o l’ Oxford Nanopore, che puntano alla riduzione dei costi e al miglioramento della qualità dei sequenziamenti, non rinunciando all’elevato throughput che caratterizza i metodi NGS.
 è stato utilizzato il metodo Sanger. (chain termination method, dideoxynucleotides ddNTPs) Questa tecnica nel corso degli anni è stata perfezionata ed automatizzata fino a raggiungere sequenze, o reads, lunghe fino a mille paia di basi, e fino ad un totale di trecento (384) sequenze per esperimento. Il metodo Sanger richiede però lunghi tempi di preparazione dei campioni (clonazione) e la quantità di paia di basi sequenziate è relativamente bassa. A partire dal 2004 sono state introdotte le tecnologie di sequenziamento chiamate Next Generation Sequencing, o NGS, sviluppate da compagnie come Roche/454 , Applied Biosystems/Solid o Illumina/Solexa. La caratteristiche principali di queste tecnologie sono la brevità delle reads sequenziate, in un range tra 35 e 1000 paia di basi a seconda della macchina utilizzata, e l’alto parallelismo automatizzato dei processi di sequenziamento, che permette di ottenere un’enorme quantità di basi sequenziate per ogni esperimento. Per questa caratteristica ci si riferisce al Next generation Sequencing anche con il termine di Massive Parallel Sequencing. Confrontando con il metodo della prima generazione vediamo come il numero di paia di basi sequenziate è da uno a due ordini di grandezza superiore. Già dallo scorso anno sono state presentate nuove tecnologie che rappresentano un’ulteriore evoluzione dei metodi NGS, come l’Ion Torrent della Life Technologies o l’ Oxford Nanopore, che puntano alla riduzione dei costi e al miglioramento della qualità dei sequenziamenti, non rinunciando all’elevato throughput che caratterizza i metodi NGS.")
28
Scopi dell’Analisi di Sequenziamento
GENOME TRASCRIPTOME DE-NOVO RE SEQUENCING LONG SHORT AMPLICON EXOME WHOLE GENOME CODING NO CODING miRNA OTHER Aspetti positivi Grande mole di dati prodotti; Identificazione cause malattie sconosciute o poco conosciute; Applicazione delle conoscenze acquisite (farmacogenomica). Limiti Necessità di server capienti; Costruzione di strutture bioinformatiche complesse; Necessità di database integrati; Diverse domande biologiche.
. Limiti. Necessità di server capienti; Costruzione di strutture bioinformatiche complesse; Necessità di database integrati; Diverse domande biologiche.")
29
RNA-seq for Reverse Engineering of the genome state
Cells/Biosamples Library preparation Sequencing Computational analysis for reverse engineering

30
Genoma < 3% del genoma umano codifica proteine
Evidenze recenti ottenute con genomic-tiling array e sequenziamento del trascrittoma hanno mostrato che >70% del genoma è trascritto in maniera pervasiva in RNA codificante ( mRNA) Moltissimi prodotti trascrizionali sono RNA, piccoli e lunghi, con scarsissimo potenziale codificante La maggior parte del DNA eucariotico trascritto è non codificante La production phase di ENCODE ha mostrato che >80% del genoma è biologicamente attivo e funzionale (ruolo regolativo per la maggior parte delle sequenze)
 Moltissimi prodotti trascrizionali sono RNA, piccoli e lunghi, con scarsissimo potenziale codificante. La maggior parte del DNA eucariotico trascritto è non codificante. La production phase di ENCODE ha mostrato che >80% del genoma è biologicamente attivo e funzionale (ruolo regolativo per la maggior parte delle sequenze)")
31
Il trascrittoma non-codificante
Le molecole di RNA possono contemporaneamente contenere informazione di sequenza e possedere plasticità strutturale Gli RNA possono sia interagire con DNA ed altri RNA per appaiamento delle basi complementari, sia fornire siti di legame per proteine RNA non codificanti noti da molto tempo: rRNA e tRNA nella traduzione snRNA e snoRNA nel processamento degli mRNA ribozimi Ipotesi del “mondo ad RNA”

32
DNA RNA mRNA ncRNA proteins
Transcription <3% of the genome is important since transcribed/coding abundant “junk DNA” RNA Processing mRNA ncRNA Translation proteins

33
RNA transcripts/precursors
>70% transcribed in “dark matter” DNA Transcription Transcription Alternative TSSs Processing RNA transcripts/precursors Splicing Nuclear export Processing Polyadenylation Silencing Editing Editing Turn-over Turn-over Trans-splicing Sequestration Sequestration mRNA Da transcibed dark matter a increase appreciation of the biological role or RNAs and integractions thereof Concept of “transcribed dark matter”, i.e. transcripts with unknown function and meaning ncRNA Translation miRNAs snRNAs lncRNAs proteins piRNAs snoRNAs circRNAs siRNAs tRFs ? Diverse functional roles for ncRNAs uncovered

34
Primary Databases: Databases consisting of data derived experimentally such as nucleotide sequences and three dimensional structures are known as primary databases. Secondary Databases: Those data that are derived from the analysis, treatment or integration of primary data such as secondary structures, hydrophobicity plots, and domain are stored in secondary databases.

35
DATABASE DI SEQUENZE NUCLEOTIDICHE
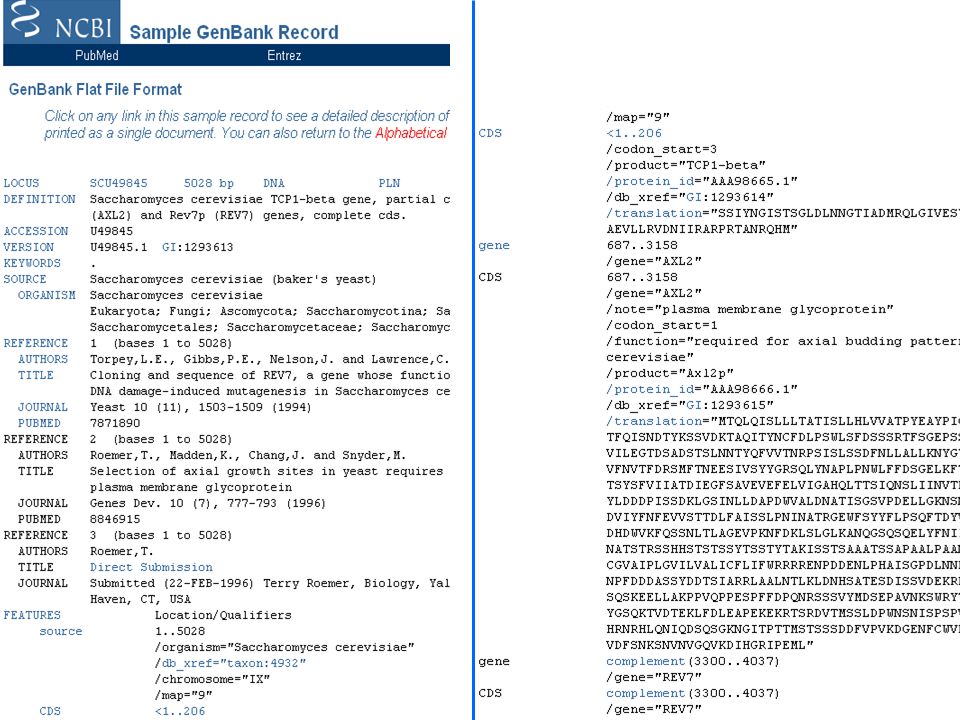
DATABASE PRIMARI DATABASE DI SEQUENZE NUCLEOTIDICHE Collezioni di singoli record, ognuno dei quali contiene un tratto di DNA o RNA con delle annotazioni. Ogni record viene anche chiamato ENTRY, e ha un codice che lo identifica univocamente (ACCESSION NUMBER). Banche dati primarie di sequenze nucleotidiche EMBL nucleotide database, ora gestita dall’EBI (1980) EMBL = European Molecular Biology Laboratory (Heidelberg) EBI = European Bioinformatics Institute (Hinxton, UK) GenBank = banca dell NIH gestita dal NCBI (1982) NIH = National Institutes of Health (Stuttura USA) NCBI = National Center for Biotechnology Information, Bethesda, Maryland DDBJ = banca DNA giapponese (1986) DDBJ = DNA DataBase of Japan SCAMBIO DI DATI Nel 1988, i gruppi responsabili dei 3 database si sono organizzati nell’International Collaboration of DNA Sequence Databases per utilizzare un formato comune e scambiarsi giornalmente le sequenze.
. Banche dati primarie di sequenze nucleotidiche EMBL nucleotide database, ora gestita dall’EBI (1980) EMBL = European Molecular Biology Laboratory (Heidelberg) EBI = European Bioinformatics Institute (Hinxton, UK) GenBank = banca dell NIH gestita dal NCBI (1982) NIH = National Institutes of Health (Stuttura USA) NCBI = National Center for Biotechnology Information, Bethesda, Maryland. DDBJ = banca DNA giapponese (1986) DDBJ = DNA DataBase of Japan. SCAMBIO DI DATI Nel 1988, i gruppi responsabili dei 3 database si sono organizzati nell’International Collaboration of DNA Sequence Databases per utilizzare un formato comune e scambiarsi giornalmente le sequenze.")
36
DATABASE DI SEQUENZE NUCLEOTIDICHE – GenBank
SUBMISSION DIRETTA La gran parte delle sequenze finisce in uno dei tre database perché l’autore (il laboratorio dove tale sequenza è stata ottenuta) la invia direttamente. La sequenza viene quindi inserita e il record corrispondente resta di proprietà solo di quel database, l’unico con il diritto di modificarlo. Il database che riceve la sequenza la invia poi agli altri due. ANNOTAZIONE Ci sono poi anche degli “annotatori” che prendono le sequenze dalle riviste scientifiche e le trasferiscono nel database. Problema della ridondanza GENBANK AND WGS STATISTICS Release 199 of Dec 2013 GenBank WGS Bases Sequences Bases Sequences 1, Dec … 199, Dec
 la invia direttamente. La sequenza viene quindi inserita e il record corrispondente resta di proprietà solo di quel database, l’unico con il diritto di modificarlo. Il database che riceve la sequenza la invia poi agli altri due. ANNOTAZIONE Ci sono poi anche degli annotatori che prendono le sequenze dalle riviste scientifiche e le trasferiscono nel database. Problema della ridondanza. GENBANK AND WGS STATISTICS. Release 199 of Dec GenBank WGS. Bases Sequences Bases Sequences. 1, Dec … 199, Dec")
39
DATABASE DI SEQUENZE PROTEICHE
DATABASE PRIMARI DATABASE DI SEQUENZE PROTEICHE SWISS-PROT Database di sequenze proteiche annotate, “scarsamente” ridondanti e cross-referenced Contiene TrEMBL, supplemento a SWISS-PROT costituito dalle sequenze annotate al computer, come traduzione di tutte le sequenze codificanti presenti all’EMBL TrEMBL contiene due sezioni: SP-TrEMBL, sequenze da incorporare in SWISSPROT, con AC. REM-TrEMBL, remaining (immunoglobuline, proteine sintetiche, ...), senza AC. TrEMBLnew, generato ogni settimana.
, senza AC. TrEMBLnew, generato ogni settimana.")
40
LOCUS AIL58882 140 aa linear BCT 29-AUG-2014
DEFINITION crystallin [Staphylococcus aureus]. ACCESSION AIL58882 VERSION AIL GI: DBLINK BioProject: PRJNA240091 DBSOURCE accession CP KEYWORDS . SOURCE Staphylococcus aureus ORGANISM Staphylococcus aureus Bacteria; Firmicutes; Bacilli; Bacillales; Staphylococcus. REFERENCE 1 (residues 1 to 140) AUTHORS Benson,M.A., Ohneck,E.A., Ryan,C., Alonzo,F. III, Smith,H., Narechania,A., Kolokotronis,S.O., Satola,S.W., Uhlemann,A.C., Sebra,R., Deikus,G., Shopsin,B., Planet,P.J. and Torres,V.J. TITLE Evolution of hypervirulence by a MRSA clone through acquisition of a transposable element JOURNAL Mol. Microbiol. 93 (4), (2014) PUBMED REFERENCE 2 (residues 1 to 140) AUTHORS Planet,P.J., Narechania,A., Shopsin,B. and Torres,V. TITLE Direct Submission JOURNAL Submitted (18-MAR-2014) Pediatrics, Columbia University, 650 West 168th St, New York, NY 10032, USA COMMENT Annotation was added by the NCBI Prokaryotic Genome Annotation Pipeline (released 2013). Information about the Pipeline can be found here: ##Genome-Annotation-Data-START## Annotation Provider :: NCBI Annotation Date :: 03/20/ :06:33 Annotation Pipeline :: NCBI Prokaryotic Genome Annotation Pipeline Annotation Method :: Best-placed reference protein set; GeneMarkS+ Annotation Software revision :: 2.4 (rev ) Features Annotated :: Gene; CDS; rRNA; tRNA; ncRNA; repeat_region Genes :: 2,836 CDS :: 2,729 Pseudo Genes :: 29 rRNAs :: 19 ( 5S, 16S, 23S ) tRNAs :: 59 ncRNA :: 0 Frameshifted Genes :: 23 ##Genome-Annotation-Data-END## FEATURES … FEATURES Location/Qualifiers source /organism="Staphylococcus aureus" /strain="2395 USA500" /db_xref="taxon:1280" Protein /product="crystallin" Region /region_name="IbpA" /note="Molecular chaperone (small heat shock protein) [Posttranslational modification, protein turnover, chaperones]; COG0071" /db_xref="CDD:223149" Region /region_name="alpha-crystallin-Hsps_p23-like" /note="alpha-crystallin domain (ACD) found in alpha-crystallin-type small heat shock proteins, and a similar domain found in p23 (a cochaperone for Hsp90) and in other p23-like proteins; cl00175" /db_xref="CDD:260235" CDS /locus_tag="CH51_12820" /coded_by="CP : " /inference="EXISTENCE: similar to AA sequence:RefSeq:WP_ " /note="Derived by automated computational analysis using gene prediction method: Protein Homology." /transl_table=11 ORIGIN 1 mnfnqfenqn ffngnpsdtf kdlgkqvfny fstpsfvtni yetdelyyle aelagvnked 61 isidfnnntl tiqatrsaky kseqlilder nfeslmrqfd feavdkqhit asfengllti 121 tlpkikpsne ttsstsipis //
 AUTHORS Benson,M.A., Ohneck,E.A., Ryan,C., Alonzo,F. III, Smith,H., Narechania,A., Kolokotronis,S.O., Satola,S.W., Uhlemann,A.C., Sebra,R., Deikus,G., Shopsin,B., Planet,P.J. and Torres,V.J. TITLE Evolution of hypervirulence by a MRSA clone through acquisition of. a transposable element. JOURNAL Mol. Microbiol. 93 (4), (2014) PUBMED REFERENCE 2 (residues 1 to 140) AUTHORS Planet,P.J., Narechania,A., Shopsin,B. and Torres,V. TITLE Direct Submission. JOURNAL Submitted (18-MAR-2014) Pediatrics, Columbia University, 650 West. 168th St, New York, NY 10032, USA. COMMENT Annotation was added by the NCBI Prokaryotic Genome Annotation. Pipeline (released 2013). Information about the Pipeline can be. found here: ##Genome-Annotation-Data-START## Annotation Provider :: NCBI. Annotation Date :: 03/20/ :06:33. Annotation Pipeline :: NCBI Prokaryotic Genome Annotation. Pipeline. Annotation Method :: Best-placed reference protein set; GeneMarkS+ Annotation Software revision :: 2.4 (rev ) Features Annotated :: Gene; CDS; rRNA; tRNA; ncRNA; repeat_region. Genes :: 2,836. CDS :: 2,729. Pseudo Genes :: 29. rRNAs :: 19 ( 5S, 16S, 23S ) tRNAs :: 59. ncRNA :: 0. Frameshifted Genes :: 23. ##Genome-Annotation-Data-END## FEATURES. … FEATURES Location/Qualifiers. source /organism= Staphylococcus aureus /strain= 2395 USA500 /db_xref= taxon:1280 Protein /product= crystallin Region /region_name= IbpA /note= Molecular chaperone (small heat shock protein) [Posttranslational modification, protein turnover, chaperones]; COG0071 /db_xref= CDD: Region /region_name= alpha-crystallin-Hsps_p23-like /note= alpha-crystallin domain (ACD) found in. alpha-crystallin-type small heat shock proteins, and a. similar domain found in p23 (a cochaperone for Hsp90) and. in other p23-like proteins; cl00175 /db_xref= CDD: CDS /locus_tag= CH51_12820 /coded_by= CP : /inference= EXISTENCE: similar to AA. sequence:RefSeq:WP_ /note= Derived by automated computational analysis using. gene prediction method: Protein Homology. /transl_table=11. ORIGIN. 1 mnfnqfenqn ffngnpsdtf kdlgkqvfny fstpsfvtni yetdelyyle aelagvnked. 61 isidfnnntl tiqatrsaky kseqlilder nfeslmrqfd feavdkqhit asfengllti. 121 tlpkikpsne ttsstsipis. //")
41
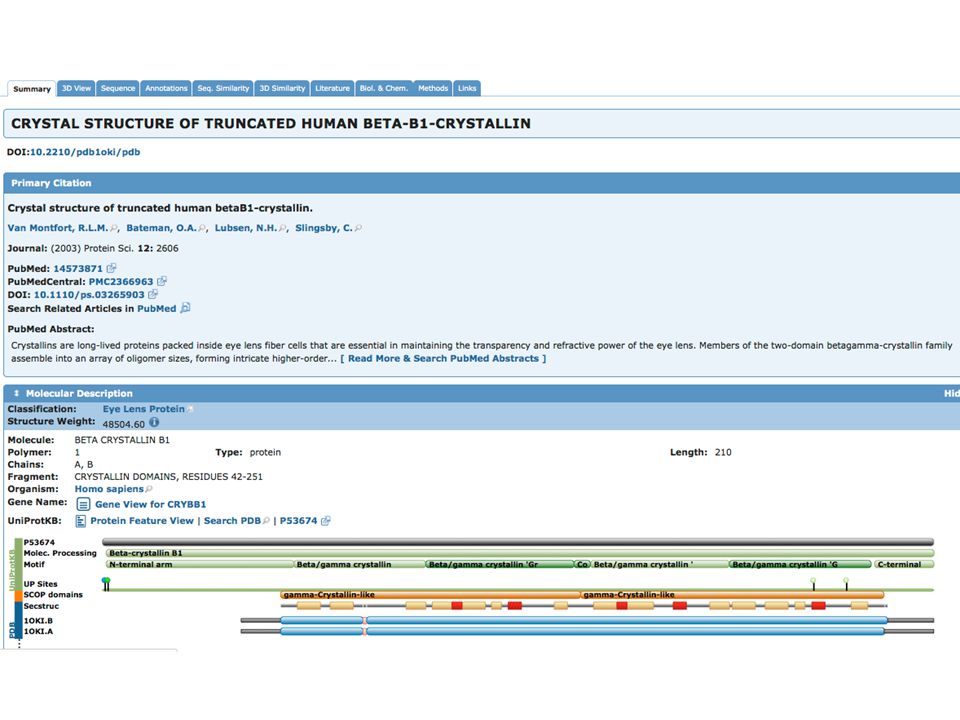
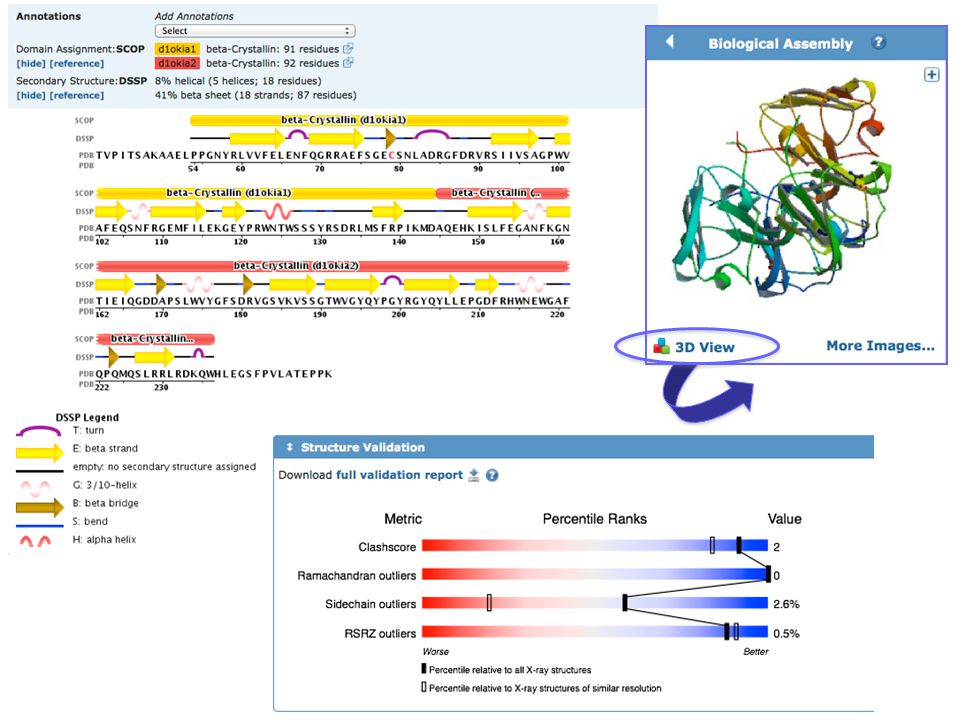
PDB Database di strutture 3-D di proteine e acidi nucleici
DATABASE PRIMARI PDB Database di strutture 3-D di proteine e acidi nucleici Dati ottenuti sperimentalmente e sottomessi direttamente dai ricercatori Fondato nel 1971

42
PDB files PDB file format
The most common format for storage and exchange of atomic coordinates for biological molecules is PDB file format PDB file format is a text (ASCII) format, with an extensive header that can be read and interpreted either by programs or by people Next slide: PDB file format
 format, with an extensive header that can be read and interpreted either by programs or by people. Next slide: PDB file format.")
43
x y z nome composto organismo autore referenze risoluzione sequenza
HEADER TRANSCRIPTION REGULATION AUG RPO RPO 2 COMPND ROP (COLE1 REPRESSOR OF PRIMER) MUTANT WITH ALA INSERTED ON 1RPO 3 COMPND 2 EITHER SIDE OF ASP 31 (INS (A-D31-A)) RPO 4 SOURCE (ESCHERICHIA COLI) RPO 5 AUTHOR M.VLASSI,M.KOKKINIDIS RPO 6 REVDAT MAY-95 1RPOA REMARK RPOA 1 REVDAT FEB-95 1RPO RPO 7 JRNL AUTH M.VLASSI,C.STEIF,P.WEBER,D.TSERNOGLOU,K.WILSON, RPO 8 JRNL AUTH 2 H.J.HINZ,M.KOKKINIDIS RPO 9 JRNL TITL RESTORED HEPTAD PATTERN CONTINUITY DOES NOT RPO 10 JRNL TITL 2 ALTER THE FOLDING OF A 4-ALPHA-HELICAL BUNDLE RPO 11 JRNL REF NAT.STRUCT.BIOL V RPO 12 JRNL REFN ASTM NSBIEW US ISSN RPO 13 REMARK RPO 14 REMARK 1 REFERENCE RPO 15 REMARK 1 AUTH M.KOKKINIDIS,M.VLASSI,Y.PAPANIKOLAOU,D.KOTSIFAKI, 1RPO 16 REMARK 1 AUTH 2 A.KINGSWELL,D.TSERNOGLOU,H.J.HINZ RPO 17 REMARK 1 TITL CORRELATION BETWEEN PROTEIN STABILITY AND CRYSTAL 1RPO 18 REMARK 1 TITL 2 PROPERTIES OF DESIGNED ROP VARIANTS RPO 19 REMARK 1 REF PROTEINS.STRUCT.,FUNCT., V RPOA 2 REMARK 1 REF 2 GENET RPOA 3 REMARK 1 REFN ASTM PSFGEY US ISSN RPO 22 REMARK RPO 29 REMARK 2 RESOLUTION ANGSTROMS RPO 30 REMARK RPO 94 REMARK 999 SEQUENCE NUMBER IS ALSO THAT FROM PDB ENTRY RPO 95 SEQRES MET THR LYS GLN GLU LYS THR ALA LEU ASN MET ALA ARG 1RPO 96 SEQRES PHE ILE ARG SER GLN THR LEU THR LEU LEU GLU LYS LEU 1RPO 97 SEQRES ASN GLU LEU ALA ASP ALA ALA ASP GLU GLN ALA ASP ILE 1RPO 98 SEQRES CYS GLU SER LEU HIS ASP HIS ALA ASP GLU LEU TYR ARG 1RPO 99 SEQRES SER CYS LEU ALA ARG PHE GLY ASP ASP GLY GLU ASN LEU 1RPO 100 ATOM N MET RPO 115 ATOM CA MET RPO 116 ATOM C MET RPO 117 ATOM O MET RPO 118 ATOM CB MET RPO 119 ATOM CG MET RPO 120 ATOM SD MET RPO 121 ATOM CE MET RPO 122 ATOM N THR RPO 123 ATOM CA THR RPO 124 ATOM C THR RPO 125 autore organismo referenze risoluzione sequenza residuo 1 residuo 2 num. residuo tipo residuo num.atomo tipo atomo x y z
 MUTANT WITH ALA INSERTED ON 1RPO 3. COMPND 2 EITHER SIDE OF ASP 31 (INS (A-D31-A)) 1RPO 4. SOURCE (ESCHERICHIA COLI) 1RPO 5. AUTHOR M.VLASSI,M.KOKKINIDIS 1RPO 6. REVDAT 2 15-MAY-95 1RPOA 1 REMARK 1RPOA 1. REVDAT 1 14-FEB-95 1RPO 0 1RPO 7. JRNL AUTH M.VLASSI,C.STEIF,P.WEBER,D.TSERNOGLOU,K.WILSON, 1RPO 8. JRNL AUTH 2 H.J.HINZ,M.KOKKINIDIS 1RPO 9. JRNL TITL RESTORED HEPTAD PATTERN CONTINUITY DOES NOT 1RPO 10. JRNL TITL 2 ALTER THE FOLDING OF A 4-ALPHA-HELICAL BUNDLE 1RPO 11. JRNL REF NAT.STRUCT.BIOL. V RPO 12. JRNL REFN ASTM NSBIEW US ISSN RPO 13. REMARK 1 1RPO 14. REMARK 1 REFERENCE 1 1RPO 15. REMARK 1 AUTH M.KOKKINIDIS,M.VLASSI,Y.PAPANIKOLAOU,D.KOTSIFAKI, 1RPO 16. REMARK 1 AUTH 2 A.KINGSWELL,D.TSERNOGLOU,H.J.HINZ 1RPO 17. REMARK 1 TITL CORRELATION BETWEEN PROTEIN STABILITY AND CRYSTAL 1RPO 18. REMARK 1 TITL 2 PROPERTIES OF DESIGNED ROP VARIANTS 1RPO 19. REMARK 1 REF PROTEINS.STRUCT.,FUNCT., V RPOA 2. REMARK 1 REF 2 GENET. 1RPOA 3. REMARK 1 REFN ASTM PSFGEY US ISSN RPO 22. REMARK 2 1RPO 29. REMARK 2 RESOLUTION. 1.4 ANGSTROMS. 1RPO 30. REMARK 1RPO 94. REMARK 999 SEQUENCE NUMBER IS ALSO THAT FROM PDB ENTRY 1RPO 95. SEQRES 1 65 MET THR LYS GLN GLU LYS THR ALA LEU ASN MET ALA ARG 1RPO 96. SEQRES 2 65 PHE ILE ARG SER GLN THR LEU THR LEU LEU GLU LYS LEU 1RPO 97. SEQRES 3 65 ASN GLU LEU ALA ASP ALA ALA ASP GLU GLN ALA ASP ILE 1RPO 98. SEQRES 4 65 CYS GLU SER LEU HIS ASP HIS ALA ASP GLU LEU TYR ARG 1RPO 99. SEQRES 5 65 SER CYS LEU ALA ARG PHE GLY ASP ASP GLY GLU ASN LEU 1RPO 100. ATOM 1 N MET RPO 115. ATOM 2 CA MET RPO 116. ATOM 3 C MET RPO 117. ATOM 4 O MET RPO 118. ATOM 5 CB MET RPO 119. ATOM 6 CG MET RPO 120. ATOM 7 SD MET RPO 121. ATOM 8 CE MET RPO 122. ATOM 9 N THR RPO 123. ATOM 10 CA THR RPO 124. ATOM 11 C THR RPO 125. autore. organismo. referenze. risoluzione. sequenza. residuo 1. residuo 2. num. residuo. tipo residuo. num.atomo. tipo atomo. x. y. z.")
45
PDB file example HEADER SYNTHETIC PROTEIN MODEL 02-JUL-90 1AL1 1AL1 2
COMPND ALPHA - 1 (AMPHIPHILIC ALPHA HELIX) AL1 3 SOURCE SYNTHETIC AL1 4 AUTHOR C.P.HILL,D.H.ANDERSON,L.WESSON,W.F.DE*GRADO,D.EISENBERG AL1 5 REVDAT JAN-95 1AL1A HET AL1A 1 REVDAT OCT-91 1AL AL1 6 JRNL AUTH C.P.HILL,D.H.ANDERSON,L.WESSON,W.F.DE*GRADO, AL1 7 JRNL AUTH 2 D.EISENBERG AL1 8 JRNL TITL CRYSTAL STRUCTURE OF ALPHA=1=: IMPLICATIONS FOR AL1 9 JRNL TITL 2 PROTEIN DESIGN AL1 10 JRNL REF SCIENCE V AL1 11 JRNL REFN ASTM SCIEAS US ISSN AL1 12 REMARK AL1 13 REMARK 1 REFERENCE AL1 14 REMARK 1 AUTH D.EISENBERG,W.WILCOX,S.M.ESHITA,P.M.PRYCIAK,S.P.HO 1AL1 15 REMARK 1 TITL THE DESIGN, SYNTHESIS, AND CRYSTALLIZATION OF AN 1AL1 16 REMARK 1 TITL 2 ALPHA-*HELICAL PEPTIDE AL1 17 REMARK 1 REF PROTEINS.STRUCT.,FUNCT., V AL1 18 REMARK 1 REF 2 GENET AL1 19 REMARK 1 REFN ASTM PSFGEY US ISSN AL1 20 REMARK AL1 21 REMARK 2 RESOLUTION. 2.7 ANGSTROMS AL1 22 REMARK AL1 23 REMARK 3 REFINEMENT. BY THE RESTRAINED LEAST SQUARES PROCEDURE OF J. 1AL1 24 REMARK 3 KONNERT AND W. HENDRICKSON (PROGRAM *PROLSQ*). THE R AL1 25 REMARK 3 VALUE IS FOR ALL DATA. THE R VALUE IS FOR ALL 1AL1 26 REMARK 3 REFLECTIONS IN THE RESOLUTION RANGE 10.0 TO 2.7 ANGSTROMS 1AL1 27 REMARK 3 WITH FOBS .GT. 2*SIGMA(FOBS). THE RMS DEVIATION FROM AL1 28 REMARK 3 IDEALITY OF THE BOND LENGTHS IS ANGSTROMS. THE RMS 1AL1 29 REMARK 3 DEVIATION FROM IDEALITY OF THE BOND ANGLE DISTANCES IS AL1 30
 1AL1 3. SOURCE SYNTHETIC 1AL1 4. AUTHOR C.P.HILL,D.H.ANDERSON,L.WESSON,W.F.DE*GRADO,D.EISENBERG 1AL1 5. REVDAT 2 15-JAN-95 1AL1A 1 HET 1AL1A 1. REVDAT 1 15-OCT-91 1AL1 0 1AL1 6. JRNL AUTH C.P.HILL,D.H.ANDERSON,L.WESSON,W.F.DE*GRADO, 1AL1 7. JRNL AUTH 2 D.EISENBERG 1AL1 8. JRNL TITL CRYSTAL STRUCTURE OF ALPHA=1=: IMPLICATIONS FOR 1AL1 9. JRNL TITL 2 PROTEIN DESIGN 1AL1 10. JRNL REF SCIENCE V AL1 11. JRNL REFN ASTM SCIEAS US ISSN AL1 12. REMARK 1 1AL1 13. REMARK 1 REFERENCE 1 1AL1 14. REMARK 1 AUTH D.EISENBERG,W.WILCOX,S.M.ESHITA,P.M.PRYCIAK,S.P.HO 1AL1 15. REMARK 1 TITL THE DESIGN, SYNTHESIS, AND CRYSTALLIZATION OF AN 1AL1 16. REMARK 1 TITL 2 ALPHA-*HELICAL PEPTIDE 1AL1 17. REMARK 1 REF PROTEINS.STRUCT.,FUNCT., V AL1 18. REMARK 1 REF 2 GENET. 1AL1 19. REMARK 1 REFN ASTM PSFGEY US ISSN AL1 20. REMARK 2 1AL1 21. REMARK 2 RESOLUTION. 2.7 ANGSTROMS. 1AL1 22. REMARK 3 1AL1 23. REMARK 3 REFINEMENT. BY THE RESTRAINED LEAST SQUARES PROCEDURE OF J. 1AL1 24. REMARK 3 KONNERT AND W. HENDRICKSON (PROGRAM *PROLSQ*). THE R 1AL1 25. REMARK 3 VALUE IS FOR ALL DATA. THE R VALUE IS FOR ALL 1AL1 26. REMARK 3 REFLECTIONS IN THE RESOLUTION RANGE 10.0 TO 2.7 ANGSTROMS 1AL1 27. REMARK 3 WITH FOBS .GT. 2*SIGMA(FOBS). THE RMS DEVIATION FROM 1AL1 28. REMARK 3 IDEALITY OF THE BOND LENGTHS IS ANGSTROMS. THE RMS 1AL1 29. REMARK 3 DEVIATION FROM IDEALITY OF THE BOND ANGLE DISTANCES IS 1AL1 30.")
46
PDB file example SEQRES ACE GLU LEU LEU LYS LYS LEU LEU GLU GLU LEU LYS GLY 1AL1 39 HET SO SULFATE ION AL1A 5 FORMUL 2 SO4 O4 S AL1 41 HELIX 1 HL1 ACE LEU AL1 42 CRYST I AL1 43 ORIGX AL1 44 ORIGX AL1 45 ORIGX AL1 46 SCALE AL1 47 SCALE AL1 48 SCALE AL1 49 ATOM C ACE AL1 50 ATOM O ACE AL1 51 ATOM CH3 ACE AL1 52 ATOM N GLU AL1 53 ATOM CA GLU AL1 54 ATOM C GLU AL1 55 ATOM O GLU AL1 56 ATOM CB GLU AL1 57 … ATOM OXT GLY AL1 144 TER GLY AL1 145 HETATM S SO AL1 146 HETATM O1 SO AL1 147 HETATM O2 SO AL1 148 HETATM O3 SO AL1 149 HETATM O4 SO AL1 150 CONECT AL1 151 CONECT AL1 152 CONECT AL1 153 CONECT AL1 154 CONECT AL1 155 MASTER AL1A 6 END AL1 157

49
Subunits view Interactive view

50
DATABASE SECONDARI

51
UniProt (Universal Protein Resource)
DATABASE SECONDARI UniProt (Universal Protein Resource) Il piu’ grande catalogo di informazioni sulle proteine. Contiene informazioni sulla sequenza e sulla funzione di proteine ed e’ ottenuto dall’insieme delle informazioni contenute in Swiss-Prot, TrEMBL e PIR.
 Il piu’ grande catalogo di informazioni sulle proteine. Contiene informazioni sulla sequenza e sulla funzione di proteine ed e’ ottenuto dall’insieme delle informazioni contenute in Swiss-Prot, TrEMBL e PIR.")
52
UniProt http://www.uniprot.org/uniprot/
UniProt Knowledgebase, due parti: Records annotati manualmente, informazioni dalla letteratura (UniProtKB/Swiss-Prot) Records risultato di analisi computazionali, in attesa di annotazione completa (UniProtKB/TrEMBL).
 Records risultato. di analisi. computazionali, in attesa di. annotazione. completa. (UniProtKB/TrEMBL).")
53
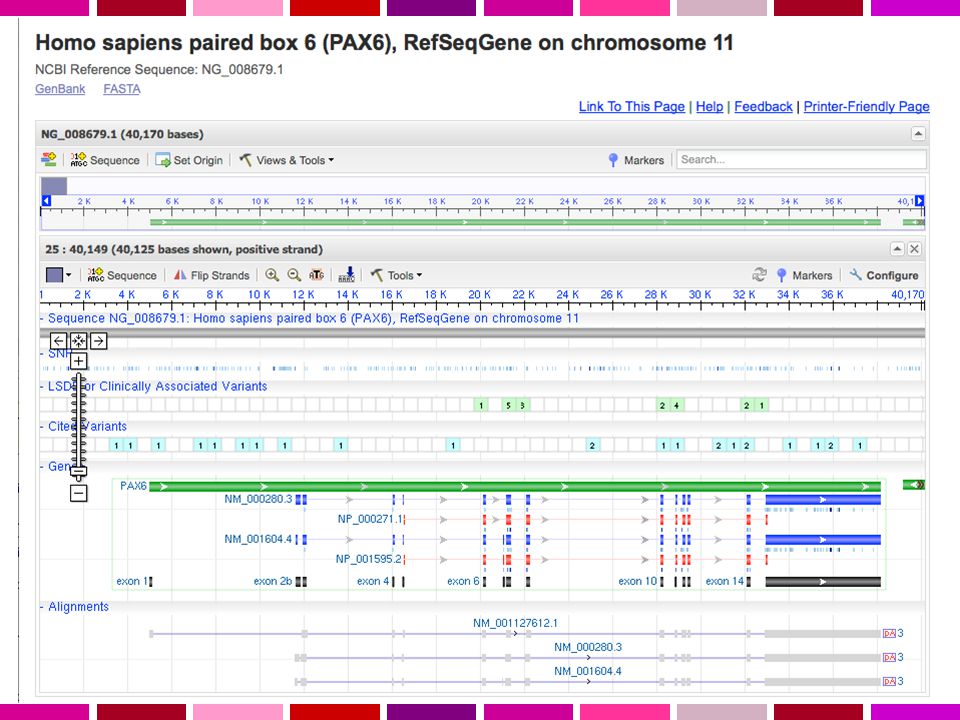
Interfaccia unificata per cercare informazioni su sequenze e loci genetici. Presenta informazioni sulla nomenclatura ufficiale, accession numbers, fenotipi, MIM numbers, UniGene clusters, omologia, posizioni di mappa e link a numerosi altri siti web. NCBI GENE

54
NCBI GENE

55
NCBI GENE

56
NCBI GENE RefSeq - Reference Sequence collection of genomic DNA, transcripts, and proteins. Distinguishing Features: non-redundancy explicitly linked nucleotide and protein sequences updates to reflect current knowledge of sequence data and biology data validation and format consistency accessions with '_' character ongoing curation by NCBI staff and collaborators, with reviewed records indicated

58
NCBI - Information retrieval system
DATABASE SECONDARI NCBI - Information retrieval system E' stato sviluppato all’NCBI (National Center for Biotechnology Information, USA) per permettere l'accesso a dati di biologia molecolare e citazioni bibliografiche. Sfrutta il concetto di “neighbouring”: possibilita' di collegare tra loro oggetti diversi di database differenti, indipendentemente dal fatto che essi siano direttamente “cross-referenced”. Tipicamente, permette l'accesso a database di sequenze nucleotidiche, di sequenze proteiche, di mappaggio di cromosomi e di genomi, di struttura 3D e bibliografici (PubMed).
 per permettere l accesso a dati di biologia molecolare e citazioni bibliografiche. Sfrutta il concetto di neighbouring : possibilita di collegare tra loro oggetti diversi di database differenti, indipendentemente dal fatto che essi siano direttamente cross-referenced . Tipicamente, permette l accesso a database di sequenze nucleotidiche, di sequenze proteiche, di mappaggio di cromosomi e di genomi, di struttura 3D e bibliografici (PubMed).")
59
PubMed

60
Bookshelf

61
Pfam Proteins contain conserved regions
Based on the conserved regions, proteins are classified into families Domains can be considered as building blocks of proteins. Some domains can be found in many proteins with different functions, while others are only found in proteins with a certain function. The presence of a particular domain can be indicative of the function of the protein.

62
Pfam The Pfam database is a large collection of protein domain families. Each family is represented by multiple sequence alignments and hidden Markov models (HMMs). HMM -> modelli probabilistici che descrivono evoluzione e conservazione di famiglie proteiche Provides links to external databases like PDB, SCOP, CATH etc.
. HMM -> modelli probabilistici che descrivono evoluzione e conservazione di famiglie proteiche. Provides links to external databases like PDB, SCOP, CATH etc.")
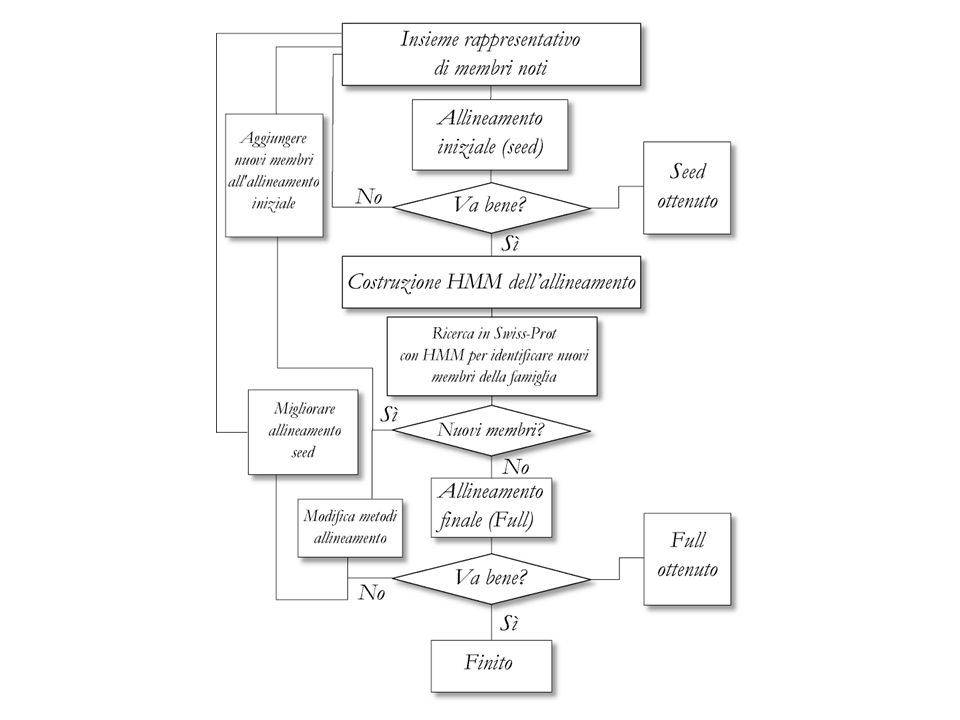
64
Pfam-A and Pfam-B Pfam-A entries are derived from the underlying sequence database (Pfamseq, built from the most recent release of UniProtKB) curated seed alignment containing a small set of representative members of the family, profile hidden Markov models (profile HMMs) built from the seed alignment, and an automatically generated full alignment.
 curated seed alignment containing a small set of representative members of the family, profile hidden Markov models (profile HMMs) built from the seed alignment, and an automatically generated full alignment.")
65
Pfam-A and Pfam-B Pfam-B families are un-annotated and of lower quality as they are generated automatically from the non-redundant clusters of the latest ADDA (Automatic Domain Decomposition Algorithm) database release. Although of lower quality, Pfam-B families can be useful for identifying functionally conserved regions when no Pfam-A entries are found.
 database release. Although of lower quality, Pfam-B families can be useful for identifying functionally conserved regions when no Pfam-A entries are found.")
66
Pfam classification Family: A collection of related protein regions
Domain: A structural unit Repeat: A short unit which is unstable in isolation but forms a stable structure when multiple copies are present Motifs: A short unit found outside globular domains Related Pfam entries are grouped together into clans; the relationship may be defined by similarity of sequence, structure or profile-HMM.

67
Pfam HMM logo Seed alignment

68
CATH Protein Structure Classification Database at UCL
Classification of proteins based on domain structures Each protein chopped into individual domains and assigned into homologous superfamilies. Hierarchial domain classification of PDB entries.

69
CATH hierarchy Class – derived from secondary structure content is assigned automatically Architecture – describes gross orientation of secondary structures, independent of connectivity (based on known structures) Topology – clusters structures according to their topological connections and numbers of secondary structures Homologous superfamily – this level groups together protein domains which are thought to share a common ancestor and can therefore be described as homologous
 Topology – clusters structures according to their topological connections and numbers of secondary structures. Homologous superfamily – this level groups together protein domains which are thought to share a common ancestor and can therefore be described as homologous.")
70
Class, C-level mainly-alpha, mainly-beta and alpha-beta (including alternating alpha/beta structures and alpha+beta structures) plus a fourth class with low secondary structure content. Architecture, A-level Overall shape; ignores the connectivity between the secondary structures. Assigned manually using literature for well-known architectures (e.g the beta-propellor or alpha four helix bundle) as reference. Topology (Fold family), T-level Structures are grouped into fold families at this level depending on both the overall shape and connectivity of the secondary structures. This is done using the structure comparison algorithm SSAP.
 plus a fourth class with low secondary structure content. Architecture, A-level. Overall shape; ignores the connectivity between the secondary structures. Assigned manually using literature for well-known architectures (e.g the beta-propellor or alpha four helix bundle) as reference. Topology (Fold family), T-level. Structures are grouped into fold families at this level depending on both the overall shape and connectivity of the secondary structures. This is done using the structure comparison algorithm SSAP.")
71
CATH – dominio maggiore serina idrossimetiltransferasi umana

72
SCOP Structural Classification of Proteins
Description of structural and evolutionary relationships between all the proteins with known structures Uses the PDB entries Search using keywords or PDB identifiers

73
Hierarchy in SCOP While the four major levels of CATH are class, architecture, topology and homologous superfamily SCOP uses: Class (all α, all β, α/β, α + β) Fold Superfamily Family Species SCOP database is mainly based on expert knowledge, while CATH grounds more on automation one with mostly parallel sheet(s), called alpha/beta, and one with mostly antiparallel sheet(s), called alpha + beta
 Fold. Superfamily. Family. Species. SCOP database is mainly based on expert knowledge, while CATH grounds more on automation. one with mostly parallel sheet(s), called alpha/beta, and one with mostly antiparallel sheet(s), called alpha + beta.")
74
What about Genomic databases
What about Genomic databases? Saranno trattati nella parte del corso riguardante la Genomica













 Piccole molecole di RNA (20-22 nt)>")









