Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
UNIVERSITA’ DI MILANO-BICOCCA LAUREA MAGISTRALE IN BIOINFORMATICA
Corso di BIOINFORMATICA: TECNICHE DI BASE Prof. Giancarlo Mauri Lezione 10 Evoluzione Molecolare e Analisi Filogenetica

2
Introduzione Problema: Struttura usata per rappresentare l’evoluzione:
studio della storia evolutiva di un insieme di specie Struttura usata per rappresentare l’evoluzione: albero evolutivo o filogenesi Struttura ad albero in cui le foglie sono etichettate dalle specie esistenti, i nodi interni dalle specie progenitrici dato un insieme di specie costruire un albero evolutivo In genere struttura dell’albero e specie progenitrici sono incognite

3
Albero evolutivo AAAGGTACC AAATGTACC AAATGTACC AAATGTGCC AAATGTGCC
G T mutation AAATGTACC A G mutation AAATGTACC AAATGTGCC TAATGTGCC AAATGTGCC A T mutation

4
I passi 1. Allineamento 2. Modello di sostituzione
3. Costruzione dell’albero 4. Valutazione dell’albero

5
Allineamento Scelta delle procedure di allineamento
Dipendenza dal computer nulla, parziale o completa Richiamo della filogenia assente, a priori o ricorsivo Stima dei parametri di allineamento a priori, dinamica o ricorsiva Possibile allineamento rispetto a strutture superiori Ottimizzazione matematica statistica o non statistica Estrazione di un insieme di dati filogenetici dall’allineamento trattamento degli indels

6
Modello di sostituzione
Matrici di sostituzione tra basi Simmetriche (reversibilità nel tempo) o no Stazionarie o no Tassi di sostituzione tra siti eterogenei Esempio: terzo codone più variabile dei primi due
 o no. Stazionarie o no. Tassi di sostituzione tra siti eterogenei. Esempio: terzo codone più variabile dei primi due.")
7
Costruzione dell’albero filogenetico
Metodi basati sulla distanza L’istanza del problema è un insieme di specie e delle distanze evolutive tra esse (Matrice delle distanze) L’obiettivo è costruire un albero che rispetti le distanze date
 L’obiettivo è costruire un albero che rispetti le distanze date.")
8
Distanza genetica tra sequenze omologhe
Numero di sostituzioni per sito Sono sottostimate (sostituzioni convergenti, retromutazioni) ACTGAACGTAACGC C->T->A A->T->A AATGGACGTAACGC TCTGGACGTAACGC
 ACTGAACGTAACGC. C->T->A. A->T->A. AATGGACGTAACGC. TCTGGACGTAACGC.")
9
Unweighted Pair Group Method with Arithmetic mean (Sokal e Michener 1958)
Funziona per velocità circa costanti nelle diverse linee evolutive: relazione lineare tra distanza e tempo di divergenza Usa un algorimo di clusterizzazione sequenziale iterativo Collega le sequenze più vicine a un antenato comune Sostituisce le due sequenze col padre Itera la procedura fino ad avere un solo elemento (radice)
")
10
UPGMA (Sokal, Michener, 1958)
Initialize Ci = {si}, for all i. Repeat until one cluster left: Find two clusters Ci, Cj with mini=1,..,n;j=1,…,n dij=(dpq)/|Ci||Cj|, pCi, qCj Define node k with i,j as children, edge weight dij Form cluster k, remove i,j clusters. 0.1 0.4 Problem of UPGMA
/|Ci||Cj|, pCi, qCj. Define node k with i,j as children, edge weight dij. Form cluster k, remove i,j clusters Problem. of UPGMA.")
11
UPGMA - Esempio A B C B dAB C dAC dBC D dAD dBD dCD
Sia dAB il valore più piccolo; A e B vengono raggruppate e il punto di biforcazione posizionato alla distanza dAB/2

12
UPGMA - Esempio AB C C d(AB)C D d(AB)D dCD
ove d(AB)C = (dAC+dBC) /2 e d(AB)D = (dAD+dBD) /2. Sia ora d(AB)C il valore più piccolo; C è raggruppata con AB con punto di biforcazione a distanza d(AB)C/2. Infine si raggruppa con D e la radice è posta a distanza d(ABC)D = [(dAD+dBD+dCD)/3] /2
C = (dAC+dBC) /2 e d(AB)D = (dAD+dBD) /2. Sia ora d(AB)C il valore più piccolo; C è raggruppata con AB con punto di biforcazione a distanza d(AB)C/2. Infine si raggruppa con D e la radice è posta a distanza d(ABC)D = [(dAD+dBD+dCD)/3] /2.")
13
UPGMA - Esempio A B dAB/2 C d(AB)C/2 D d(ABC)D /2
C/2 D d(ABC)D /2")
14
Neighbor Joining (Saitou, Nei, 1987)
Ricostruisce l’albero senza radice che minimizza la somma delle lunghezze dei rami Neighbors: coppia di sequenze, singole o composite, connesse attraverso un singolo nodo interno A B C D E

15
Neighbor Joining (Saitou-Nei, 1987)
Initialize: T={sequences}, L=T Choose i,jL such that dij-ri-rj minimized. Rest similar to UPGMA with similar modification on edge weights to k. Here, ri, rj are the average distances from i,j to other nodes in L – to compensate long edges.

16
Neighbor joining - Esempio
Situazione iniziale:

17
Neighbor joining - Esempio
Tra le n(n-1)/2 diverse coppie si cerca quella che minimizza la somma delle lunghezze dei rami nell’albero seguente:
/2 diverse coppie si cerca quella che minimizza la somma delle lunghezze dei rami nell’albero seguente:")
18
Neighbor joining - Esempio
Si itera la procedura sulla nuova stella con n-1 foglie ottenuta sostituendo ai due neighbors trovati la loro combinazione

19
Costruzione dell’albero filogenetico
Metodi basati sulle sequenze Istanza del problema : insieme di sequenze biologiche appartenenti a diverse specie Output: albero evolutivo (con i nodi interni etichettati dalle sequenze progenitrici) di costo minimo Punteggio di un arco := punteggio dell’allineamento ottimale delle sequenze associate ai nodi dell’arco Punteggio dell’albero := somma dei punteggi degli archi Caso particolare: la struttura dell’albero viene data. Ricerca sequenze progenitrici. Anche questo caso è difficile.
 di costo minimo. Punteggio di un arco := punteggio dell’allineamento ottimale delle sequenze associate ai nodi dell’arco. Punteggio dell’albero := somma dei punteggi degli archi. Caso particolare: la struttura dell’albero viene data. Ricerca sequenze progenitrici. Anche questo caso è difficile.")
20
Maximum parsimony (MP - Eck, Dayhoff 66)
Rasoio di Occam: La miglior spiegazione dei dati è la più semplice Si trova l’albero che spiega le differenze osservate col minor numero di sostituzioni Metodo qualitativo; determina la topologia dell’albero, non la lunghezza dei rami Molto lento. Usa branch and bound

21
MP Siti informativi: favoriscono alcuni alberi rispetto ad altri
In generale, contengono almeno due nucleotidi ciascuno dei quali è presente in almeno due sequenze MP è molto usato per la sua semplicità; è inadeguato per sequenze nucleotidiche, attendibile come analisi preliminare per le proteine Genera molti alberi equivalenti

22
Maximum Likelihood (ML, Felsenstein 81)
Cerca il modello evolutivo, albero compreso, che ha la massima verosimiglianza rispetto alla produzione delle sequenze osservate

23
Maximum Likelihood Modello di Jukes-Cantor (1969) : uguale probabilità di sostituzione (1 parametro ) Modello di Kimura (1980) (2 parametri): diversi tassi di sostituzione ( e ) da purina (A,G) a purina o da pirimidina (C,T,U) a pirimidina Processo molto lento, per la necessità di eseguire una ricerca esaustiva su tutti gli alberi Risultati migliori di MP nelle simulazioni
 (2 parametri): diversi tassi di sostituzione ( e ) da purina (A,G) a purina o da pirimidina (C,T,U) a pirimidina. Processo molto lento, per la necessità di eseguire una ricerca esaustiva su tutti gli alberi. Risultati migliori di MP nelle simulazioni.")
24
ML - Esempio t1 1 a t5 5 b 2 b t2 a t3 3 c 6 g t6 4 d t4
6 5 a b g t1 t2 t3 t4 t5 t6 L = SaSbSgpa Pab(t5) Pag(t6) Pba(t1) Pbb(t2) Pgc(t3) Pgd(t4)
 Pag(t6) Pba(t1) Pbb(t2) Pgc(t3) Pgd(t4)")
25
ML - Esempio Problema della determinazione di Pij(t)
Necessità di considerare diverse topologie e diverse lunghezze dei rami.

26
Metodo dei quartetti Per ogni quattro sequenze si costruisce un albero di 4 nodi (quartetto), ad esempio usando ML Si costruisce poi un grande albero formato dalla (maggior parte di) questi piccoli alberi. Questo passo è NP-difficile Un nuovo approccio: correzione dei dati
 questi piccoli alberi. Questo passo è NP-difficile. Un nuovo approccio: correzione dei dati.")
27
Quartetti e Correzione
Albero originale d b c a b e c d a d a b e e b b d a d e c correzione a c e c errore d e

28
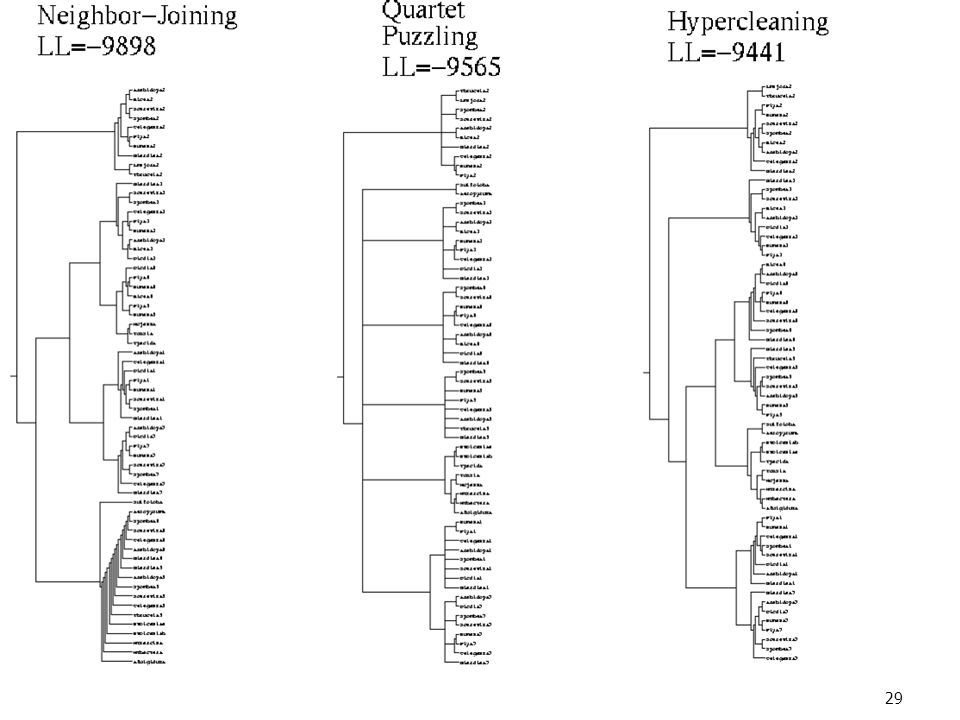
Il Software HyperCleaning
Per meno di 30 taxa, HyperCleaning è confrontabile con fastDNAml (che usa il punteggio di maximum likehood), e si comporta meglio di NJ. Per più di 30 taxa, i metodi ML e MP puri richiedono giorni e producono risultati scadenti. HyperCleaning si comporta bene, con punteggi migliori.
, e si comporta meglio di NJ. Per più di 30 taxa, i metodi ML e MP puri richiedono giorni e producono risultati scadenti. HyperCleaning si comporta bene, con punteggi migliori.")
30
Valutazione degli alberi: bootstrap (Efron 79)
Data la matrice di allineamento A di N sequenze lunghe L si generano n (es, n=100) allineamenti simulati : Per j da 1 a L, si estrae un numero casuale r tra 1 e L e si pone la j-esima colonna di Ak uguale alla r-esima di A si costruiscono gli alberi filogenetici Si attribuisce a ogni nodo un coefficiente di significatività pari alla percentuale di simulazioni che lo supportano
 allineamenti simulati : Per j da 1 a L, si estrae un numero casuale r tra 1 e L e si pone la j-esima colonna di Ak uguale alla r-esima di A. si costruiscono gli alberi filogenetici. Si attribuisce a ogni nodo un coefficiente di significatività pari alla percentuale di simulazioni che lo supportano.")
31
Confronto tra filogenesi
Tutti i metodi visti sono NP-hard E’ possibile costuire alberi approssimanti e confrontarli per ottenere un albero migliore

32
Problemi di confronto L’istanza dei problemi di confronto è un insieme di alberi evolutivi. Esistono vari problemi di confronto MAST MIT

33
MIT Maximum Isomorphic Subtree
L’obiettivo è individuare un sottoalbero S’ tali che gli alberi ristretti a S’ siano tutti isomorfi. Due alberi sono isomorfi se qualunque coppia di foglie ha uguale distanza in entrambi gli alberi. Nel caso gli alberi siano pesati si ha un nuovo problema: MWT (Maximum Weighted Subtree)
")
34
MAST Maximum agreement subtree
L’obiettivo del problema è individuare il massimo sottoinsieme di specie S’ per cui gli alberi ristretti all’insieme S’ sono omomorfi. Due alberi sono omomorfi se risultano isomorfi a meno di nodi di grado 1.

35
Complessità dei problemi di confronto
I problemi di confronto sono NP-hard già su tre alberi Inoltre non sono facilmente trattabili per l’approssimazione

36
Software filogenetico
PHYLIP PROTDIST PROTPARS DNADIST DNAML fastDNAml PAUP

Presentazioni simili


, trovare un cammino minimo.>")


>")



>")






 |cfc|=2 prima e dopo (b) |cfc|=2 prima e |cfc|=1 dopo>")


