Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Rappresenta la più comune malattia monogenica diffusa nel mondo
La talassemia Rappresenta la più comune malattia monogenica diffusa nel mondo Portatori sani

2
La Talassemia (o Anemia Mediterranea o Morbo di Cooley)
é una malattia ereditaria del sangue, a trasmissione autosomica recessiva, con alta prevalenza in alcune aree del mondo, in particolare nel bacino del Mediterraneo (Italia, Grecia, Turchia, Cipro, Marocco, Arabia Saudita) e nel Sud-Est asiatico (India, Vietnam, Cambogia, Giappone).
 e nel Sud-Est asiatico. (India, Vietnam, Cambogia, Giappone).")
3
Diffusione e frequenza della talassemia in Italia
2.4% Portatori Sani Malati 7000 7.5% 12% 2% 0.8% 5% 2.25% 8% 13% 5.9% 3% 9% 7%

4
Diffusione della talassemia in Sicilia
1042 casi talassemia major 395 casi talassemia intermedia 287 casi talassodrepanocitosi portatori sani di talassemia 6% portatore di trait talassemico 10% Sud Est dell’isola 3-4 % Nord-Ovest

6
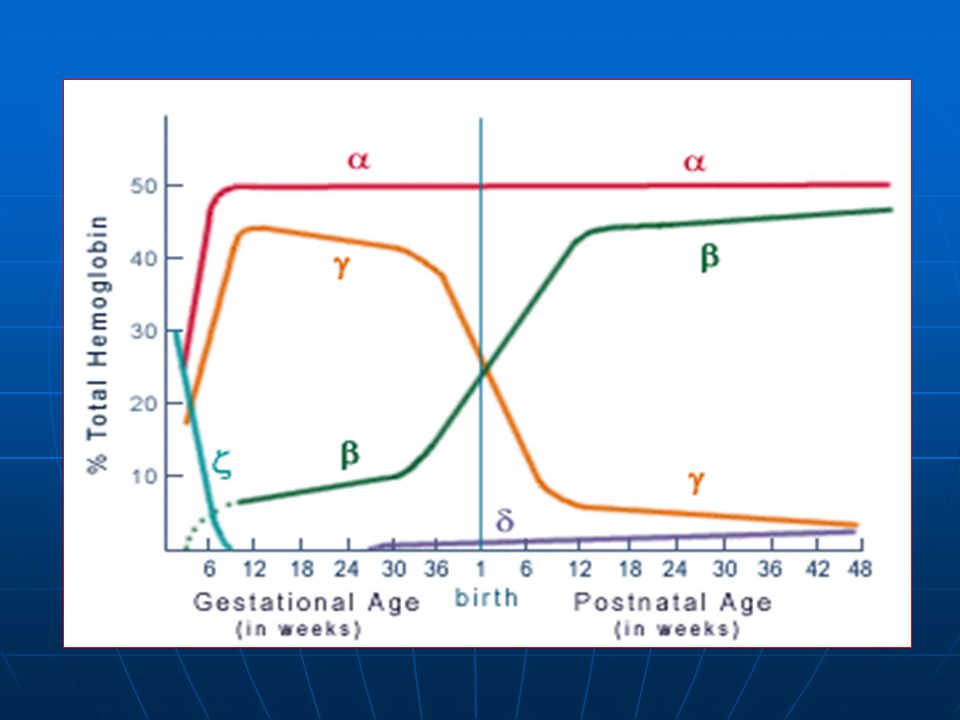
è caratterizzata da sintesi ridottissima o del tutto
La Talassemia è caratterizzata da sintesi ridottissima o del tutto assente di catene b-globiniche (cr. 11) e conseguente grave anemia Hb GR
 e conseguente grave anemia. Hb. GR.")
7
Talassemia major Insorgenza: (4-6 mesi di età)
Sintomi clinici alla presentazione: Pallore Febbricola Irritabilità Turbe dell’alvo Episodi infettivi ricorrenti Epatoslenomegalia

8
Quadro Ematologico Hb 4-6 g/dl Globuli rossi 2000000 mmc
Eritroblastemia Anisopoichilocitosi Resistenza globulari aumentate Iperplasia eritroblastica midollare Hb F 70-90% Hb A2 > 3.5%

9
Talassemia Major: striscio di sangue periferico

10
Diagnosi ematologica della talassemia major
Esame emocromocitometrico HPLC (high performance liquid chromatography)
")
11
Patogenesi della beta talassemia major

12
Quali sono le conseguenze
della Talassemia? Inadeguata produzione delle catene globiniche-->ipocromia e microcitosi accumulo non bilanciato di altre catene globiniche-->precipitati negli eritroblasti eritropoiesi inefficace ridotto numero di eritrociti maturi distruzione periferica con anemia emolitica

13
Foto storiche della espressività della malattia
Foto attuali nei paesi sottosviluppati Foto attuali nei paesi dove si applicano i protocolli di tratttamento

14
Trattamento: terapia di supporto
Terapia trasfusionale Terapia ferrochelante Induttori dell’Hb F (HU)
")
15
Terapia Trasfusionale
Correzione anemia Soppressione eccesso di eritropoiesi Inibizione assorbimento intestinale di Fe legato all’anemia

16
Regimi trasfusionali nella Talassemia Major
Anni Hb pretrasfusionale 1955 – < 6 g/dl 1961 (Orsini e coll.) > 8 g/dl 1969 (Wolman e Ortolani) ,5 – 10 g/dl 1980 (Propper) ,5 – 12 g/dl 1995 – ,5 ± 0,4 g/dl
 > 8 g/dl (Wolman e Ortolani) 9,5 – 10 g/dl (Propper) 11,5 – 12 g/dl – ,5 ± 0,4 g/dl.")
17
Terapia Ferrochelante
Desferoxamina (DFO): per via sottocutanea ad infusione lenta Deferiprone (L1): per via orale ICL 670 A: per via orale GT56-252: Un chelante orale in via di sperimentazione
: per via sottocutanea ad infusione lenta. Deferiprone (L1): per via orale. ICL 670 A: per via orale. GT56-252: Un chelante orale in via di sperimentazione.")
18
Complicanze nella talassemia major
Epatiche : epatiti acute e croniche. Emocromatosi Cardiache: Pericarditi, Miocardioptie, Insufficienza Cardiaca Endocrine: Ipogonadismo, Diabete mellito, Osteoporosi, Ipotiroidismo, Ipoparatiroidismo.

19
Complicanze nella talassemia major
Emolisi: Alloimmunizzazione anti eritrocitaria, anti Rh Infezioni Legate al trattamento Ferrochelante con DFO ed L1

20
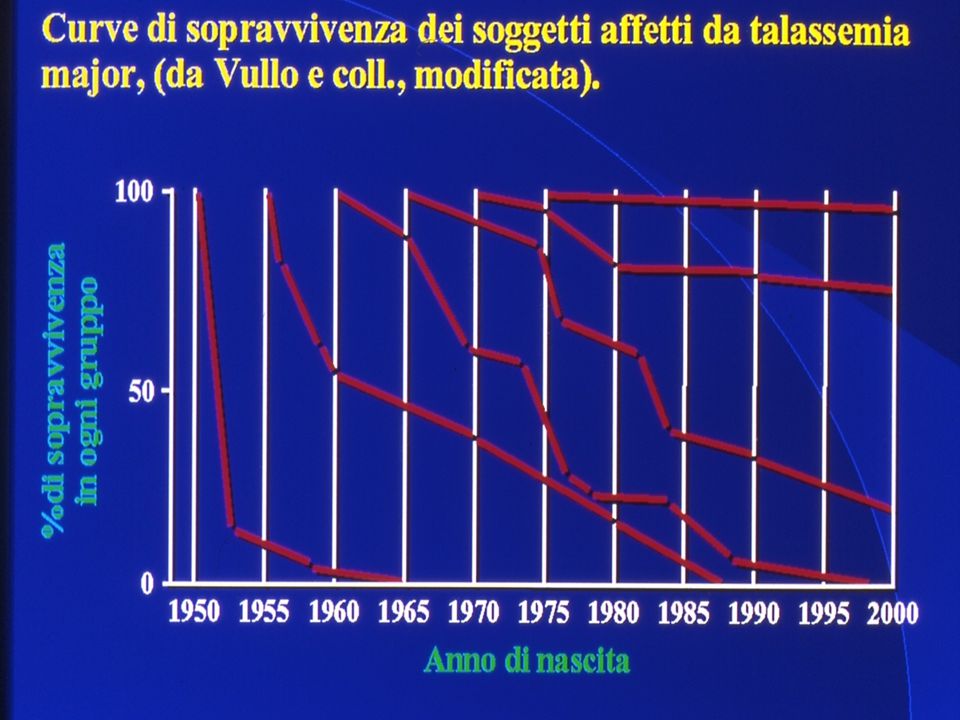
Progress in treatment

22
DISTRIBUZIONE IN ANNI DEI CASI REGISTRATI
Talassemia Major 3 2 5 2 N° pazienti 1 5 1 5 < 5 6-10 11-15 16-20 21-25 26-30 31-35 36-40 41-45 > 46 ANNI Assessorato Regionale Sanità D.E.R. - Gruppo 48 - dati R.E.S.T.E. - Agosto 1999

23
Qualità della vita Inserimento scolastico Attività lavorativa
Vita affettiva Immagine di se stesso

25
Terapia non convenzionale:
OBIETTIVO GUARIGIONE Sostituire il compartimento emopoietico alterato con un patrimonio di cellule staminali ottenuto da un donatore sano capace di ricostituire il sistema emopoietico ed immunitario del ricevente

26
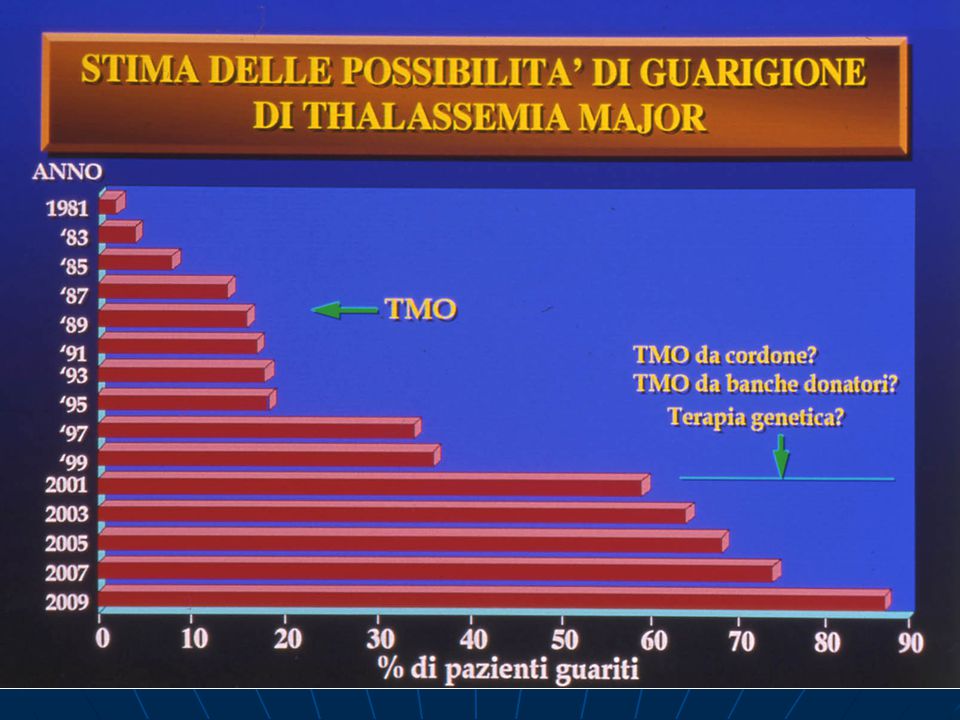
Trapianto di midollo osseo(TMO)
Trapianto di cellule staminali Terapia genica

27
Fonti di cellule staminali
Midollo osseo Sangue periferico Cordone ombelicale Tessuti fetali Embrioni

28
Trapianto di midollo osseo
I geni determinanti la compatibilità tissutale sono raggruppati come Complesso Maggiore di Istocompatibilità (MHC) – HLA Requisito fondamentale per il TMO è la disponibilità di un donatore HLA-compatibile
 – HLA. Requisito fondamentale per il TMO è la disponibilità di un donatore HLA-compatibile.")
30
Probabilità di trovare un donatore HLA - identico
Nella famiglia: 25% Nei donatori geneticamente estranei: a) numero di donatori disponibili b) eterogeneità etnica della popolazione c) frequenza degli aplotipi
 numero di donatori disponibili. b) eterogeneità etnica della popolazione. c) frequenza degli aplotipi.")
31
TMO allogenico Da donatore singenico (gemello monocoriale)
Da donatore familiare HLA identico Da donatore familiare HLA non - identico Da donatore HLA identico non - correlato

32
Fig.2 Lucarelli

33
(Per l’identificazione
PREVENZIONE Screening (Per l’identificazione del portatore sano) Diagnosi prenatale (per una scelta consapevole ) Campagne di informazione Consulenza genetica
 Diagnosi prenatale. (per una scelta. consapevole ) Campagne di. informazione. Consulenza. genetica.")
34
Modalità di trasmissione della talassemia

35
Classificazione clinica
Portatore sano Talassemia major Talassemia intermedia

36
b-talassemia trait: striscio di sangue periferico

37
A chi è rivolta la Prevenzione?
Associazioni di Volontariato Studenti delle medie inferiori e superiori Studenti universitari Personale docente e non delle scuole coinvolte I genitori degli studenti Gli utenti dei medici di base Gli utenti dei consultori familiari (donne in età fertile ed adolescenti) Gli utenti dei poliambulatori Le coppie che frequentano i corsi prematrimoniali Gli impiegati in aziende, uffici, enti pubblici
 Gli utenti dei poliambulatori. Le coppie che frequentano i corsi prematrimoniali. Gli impiegati in aziende, uffici, enti pubblici.")
38
Screening esami di I Livello
Esame emocromocitometrico dosaggio dell’HbA2 (con HPLC) valutazione qualitativa di Hb patologiche (mediante elettroforesi) dosaggio di HbF (con HPLC) dosaggio enzimatico della Znpp e/o dosaggio ematico della ferritina
 valutazione qualitativa di Hb patologiche (mediante elettroforesi) dosaggio di HbF (con HPLC) dosaggio enzimatico della Znpp e/o dosaggio ematico della ferritina.")
39
Screening esami di II Livello
Studio dei familiari

40
Screening esami di III Livello
Studio del DNA del paziente e/o dei familiari per l’identificazione della mutazione Mutazioni più frequenti in Sicilia: -87 FrCd 6 Cd 39 IVS1:110 IVS2:1 IVS1:6 IVS1:1 IVS2:745

41
La diagnosi prenatale La diagnosi prenatale si basa
sull'identificazione diretta di mutazioni nel DNA fetale in gravidanze a rischio. Tale indagine non prevede nessun tipo di rischio né per la madre per il feto. E’ una tappa obbligatoria per l’esecuzione del trapianto in utero

42
Prelievo di sangue dai villi coriali (Villocentesi)
(VIII- X settimana di gestazione) 2) Prelievo di sangue fetale (Cordocentesi) (XVIII- XXII settimana)
 2) Prelievo di sangue fetale. (Cordocentesi) (XVIII- XXII settimana)")
43
Per saperne di più: Weatherall DJ, Phenotype-genotype relationship in monogenic disease:lessons from the thalassemias. Nat Rev Genet 2, ,2001 Borgna Pignatti C, Rugolotto S, Destefano P, et al. Survival and disease complication in talassemia major. Ann N Y Acad Sci 1998;852: Cao A, Rosatelli MC, Galanello R. et al Screening for thalassemia: a model of success. Obstet Gynecol Clin N Am 29:303-28,2002 Fischer R, Tiemann C, Nielsen P, Gabe EE, et al :Assesment of iron stores in children with transfusion siderosis by biomagnetic liver susceptometry. Am J H Ematol 1999;60:289-99 Lucarelli G, Andreani M, Angelucci E .The cure of thalassemia by bone marrow trasplantation. Blood Rev 2002 Jun;16(2):81-5 Morabito N, Lasco A, Gaudio A, Meo A, et al Bisphosfonates in the treatment of thalassemia-induced osteoporosis. Osteopor Int (2002)13:
:81-5. Morabito N, Lasco A, Gaudio A, Meo A, et al Bisphosfonates in the treatment of thalassemia-induced osteoporosis. Osteopor Int (2002)13:")
Presentazioni simili


















 (ANEMIE REFRATTERIE)>")
 per la formazione di epatociti Dr. Negri Stefano.>")
