Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
III LEZIONE Allineamento di sequenze
Allineamento globale e allineamento locale Allineamento di sequenze a coppie o multiplo Ricerca di similarita’ BLAST

2
Alessandro Coppe

3
Percentuale di omologia
RICERCA DI SIMILARITÀ SIMILARITA’ ? OMOLOGIA OMOLOGIA proprieta’ di caratteri (sequenze) dovuta alla loro derivazione dallo stesso antenato comune SIMILARITA’ “grado” di somiglianza tra 2 sequenze La similarita’ osservata tra due sequenze PUO’ indicare che esse siano omologhe, cioe’ evolutivamente correlate La similarita’ e’ una proprieta’ quantitativa, si puo’ misurare L’omologia e’ una proprieta’ qualitativa, non si puo’ misurare. La similarita’ tra sequenze si osserva, l’omologia tra sequenze si puo’ ipotizzare in base alla similarita’ osservata. Percentuale di similarita’ Ricerca di similarita’ Percentuale di omologia Ricerca di omologia
 dovuta alla loro derivazione dallo stesso antenato comune. SIMILARITA’ grado di somiglianza tra 2 sequenze. La similarita’ osservata tra due sequenze PUO’ indicare che esse siano omologhe, cioe’ evolutivamente correlate. La similarita’ e’ una proprieta’ quantitativa, si puo’ misurare. L’omologia e’ una proprieta’ qualitativa, non si puo’ misurare. La similarita’ tra sequenze si osserva, l’omologia tra sequenze si puo’ ipotizzare in base alla similarita’ osservata. Percentuale di similarita’ Ricerca di similarita’ Percentuale di omologia. Ricerca di omologia.")
4
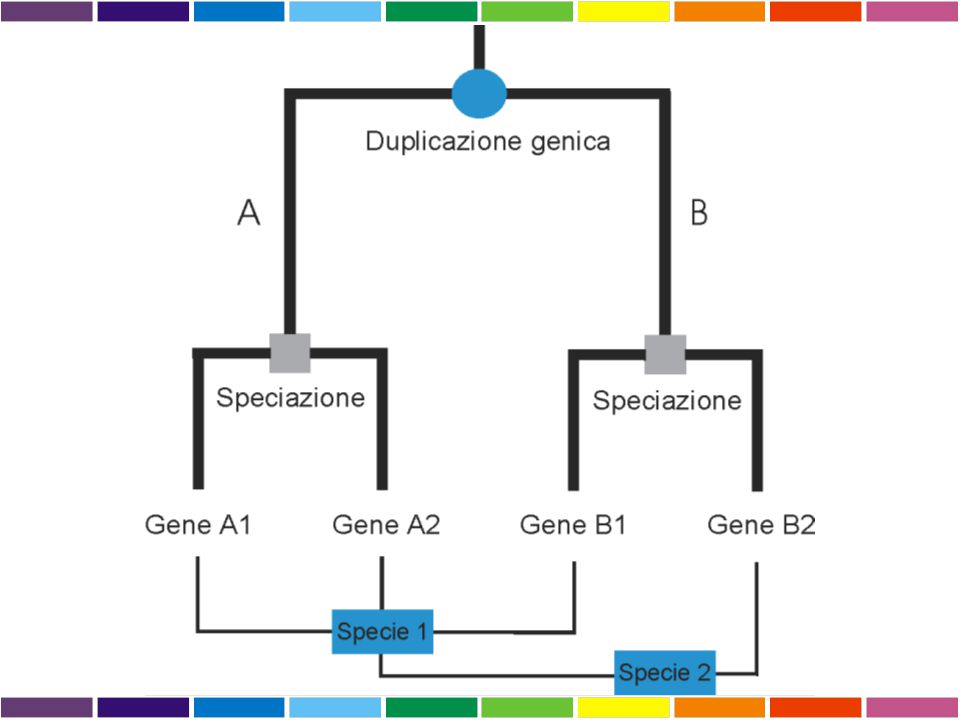
OMOLOGIA E OMOPLASIA ORTOLOGIA E PARALOGIA OMOLOGIA
Omologia similarita’ dovuta a derivazione dallo stesso antenato comune Omoplasia similarita’ dovuta a convergenza, stessa pressione selettiva su due linee evolutive puo’ condurre a caratteri simili ORTOLOGIA E PARALOGIA OMOLOGIA ANTENATO COMUNE ORTOLOGIA PARALOGIA PROCESSO DI SPECIAZIONE DUPLICAZIONE GENICA Descrivo le relazioni tra geni di una famiglia intraorganismo (paralogia) o tra diversi organismi (ortologia)
 o tra diversi organismi (ortologia)")
6
Inverso complementare
G A C T T G A T C A A G T C C T G A A C T A G T T C A G G A C T T G A Reverse A G T T C A G Complement T C A A G T C

7
Analisi di sequenze - Traduzione
Il codice genetico Senza sovrapposizione triplette codoni 20 amminoacidi e 4 nucleotidi 4, 42, 43=64 piu’ parole del necessario degenerazione (tutti i codoni hanno un significato alcuni aa sono specificati da piu’ codoni. Numero di codoni per aa: da 1 a 6 Vacillamento nella terza posizione.

8
Es. Serina Codone tRNA anticodone UCU o UCC tRNAser1 AGG + vacillamento UCA o UCG tRNAser2 AGU + vacillamento AGU o AGC tRNAser3 UCG + vacillamento Codoni di STOP: UAG, UGA e UAA Diversi codici genetici Codice genetico mitocondriale di animali AUA Met invece di Ile UGA Trp invece di STOP AGA e AGG STOP invece di Arg (UAA, UAG, AGA, AGG) Altri codici in micoplasmi, protozoi e funghi.
 Altri codici in micoplasmi, protozoi e funghi.")
9
Mascheramento 25% del genoma degli eucarioti e formata da DNA altamente ripetitivo Ripetizioni in tandem, DNA a sequenza semplice (LCR regioni a bassa complessita’) Sequenza delle ripetizioni e’ specie specifica Primati: alpha satellite 340 basi Minisatellite (6 bp) telomeri Microsatellite (unita’<4 bp, <150 ripetizioni)
 Sequenza delle ripetizioni e’ specie specifica. Primati: alpha satellite 340 basi. Minisatellite (6 bp) telomeri. Microsatellite (unita’<4 bp, <150 ripetizioni)")
10
ALLINEAMENTO DI SEQUENZE
Procedura per comparare due o piu’ sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita’ tra le sequenze stesse

11
Allineare due sequenze (proteine o acidi nucleici)
Cosa vuol dire allineare due sequenze? seq1: TCATG seq2: CATTG TCAT-G .CATTG 4 caratteri uguali 1 inserzione/delezione

12
Cosa vuol dire allineare due sequenze?
Scrivere due sequenze orizzontalmente in modo da avere il maggior numero di simboli identici o simili in registro verticale anche introducendo intervalli (gaps – inserzioni/delezioni – indels)
")
13
ALLINEAMENTO DI SEQUENZE
A COPPIE AGTTTGAATGTTTTGTGTGAAAGGAGTATACCATGAGATGAGATGACCACCAATCATTTC ||||||||||||||||||| |||||||| ||| | |||||| ||||||||||||||||| AGTTTGAATGTTTTGTGTGTGAGGAGTATTCCAAGGGATGAGTTGACCACCAATCATTTC MULTIPLO KFKHHLKEHLRIHSGEKPFECPNCKKRFSHSGSYSSHMSSKKCISLILVNGRNRALLKTl KYKHHLKEHLRIHSGEKPYECPNCKKRFSHSGSYSSHISSKKCIGLISVNGRMRNNIKT- KFKHHLKEHVRIHSGEKPFGCDNCGKRFSHSGSFSSHMTSKKCISMGLKLNNNRALLKRl KFKHHLKEHIRIHSGEKPFECQQCHKRFSHSGSYSSHMSSKKCV KYKHHLKEHLRIHSGEKPYECPNCKKRFSHSGSYSSHISSKKCISLIPVNGRPRTGLKTs

14
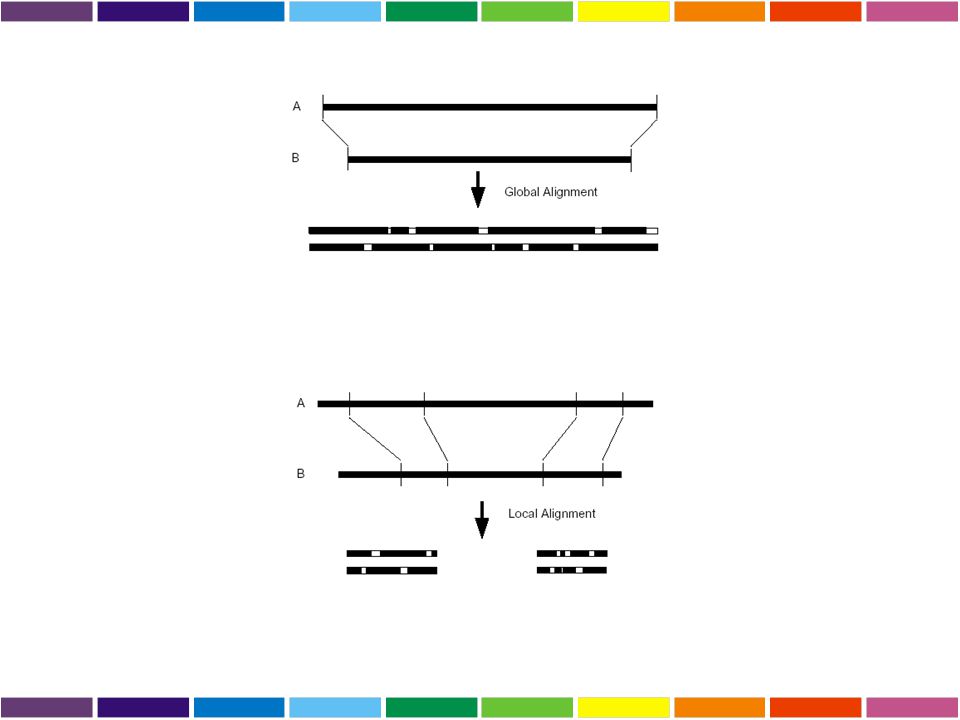
Allineamento GLOBALE o LOCALE
GLOBALE considera la similarita’ tra due sequenze in tutta la loro lunghezza LOCALE considera solo specifiche REGIONI simili tra alcune parti delle sequenze in analisi Global alignment LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||. | | | .| .| || || | || TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG Local alignment LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||||||||.|||| TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHK

16
Significato dell’allineamento
L’allineamento tra due sequenze biologiche è utile per scoprire informazione funzionale, strutturale ed evolutiva

17
Fibrosi Cistica: ereditarietà
Nei primi ani 80 è stato ipotizato che la fibrosi cistica fosse una malattia autosomica recessiva Una mutazione specifica era presente nel 70% dei pazienti con FC Similarità tra il gene che causa la FC e il gene per una ATP binding protein

18
Fibrosi Cistica e il gene CFTR :

19
Fibrosi Cistica e la proteina CFTR :
CFTR (Cystic Fibrosis Transmembrane conductance Regulator) ptoteina che agisce nella mambrana cellualre di cellule epiteliali che secernono muco
 ptoteina che agisce nella mambrana cellualre di cellule epiteliali che secernono muco.")
20
AAGGCCTAACCCCTTTGTCC
Allineamento manuale basato sulla massimizzazione del numero residui identici allineati seq1 AACCGTTGACTTTGACC Seq2 ACCGTAGACTAATTAACC AACCGTTGACT..TTGACC | ||||.|||| ||.||| A.CCGTAGACTAATTAACC Fattibile solo per poche sequenze molto brevi! Possono esistere piu’ allineamenti “equivalenti” AACCGAAGGACTTTAATC AAGGCCTAACCCCTTTGTCC AA..CCGAAGGACTTTAATC AACCGAAGGACT TTAATC || |..||...||||...| | |||.|| ||..|| AAGGCTAAACCCCTTTGTCC A AGGCCTAACCCCTTTGTC

21
+------------------- A T C A G T A
Un metodo molto semplice ed utile per la comparazione di due sequenze e’ quello della MATRICE DOTPLOT A|X X X T| X X G| X T| X X A T C A C T G T A C| X | | | | | | | A|X X X A T C A - - G T A C| X A T C A G T A

22
MISURE DI IDENTITA’ E DI SIMILARITA’
Si può misurare la similarità tra due sequenze in due diversi modi: Basato sulla percentuale di identità Basato sulla conservazione

23
MISURE DI IDENTITA’ E DI SIMILARITA’
Il modo piu’ semplice per definire le relazioni di similarita’ tra nucleotidi e’ basato solo su IDENTITA’ e DIVERSITA’. La piu’ semplice matrice di similarita’ per i nucleotidi e’ la “UNITARY SCORING MATRIX”, matrice che assegna punteggio 1 a coppie di residui identici e 0 ai mismatches. A C G T A | C | G | T | Possono esserci altri criteri per dare un peso diverso da zero a matches tra residui non identici (ad.es. Pesare in modo diverso transizioni e transversioni)
")
24
Percent Sequence Identity
The extent to which two nucleotide or amino acid sequences are invariant A C C T G A G – A G A C G T G – G C A G mismatch indel 70% identical

25
MISURE DI IDENTITA’ E DI SIMILARITA’
E’ possibile misurare la similarita’ tra aminoacidi tenendo conto delle loro proprieta’ chimico-fisiche ad. es. l’ acido glutammico e’ piu’ simile all’acido aspartico che alla fenilalanina Un altro modo per misurare la similarita’ tra aminoacidi e’ fondato sulle frequenze osservate di specifiche sostituzioni aminoacidiche in opportuni gruppi di allineamenti. La similarita’ tra due specifici aminoacidi, diciamo A e G, e’ proporzionale alla frequenza con cui si osserva la sostituzione A->G. Le MATRICI DI SOSTITUZIONE piu’ conosciute ed utilizzate sono le matrici PAM (o Dayhoff Mutation Data (MD) Matrices) e le matrici BLOSUM.
 Matrices) e le matrici BLOSUM.")
26
Matrici di sostituzione
Le matrici di sostituzione si basano su evidenze biologiche Gli allineamenti possono essere pensati come sequenze che differiscono a causa di mutazioni Alcune di queste mustazioni hanno effetti trascurabili sulla struttura/funzione della proteina

27
Esempio di matrice di sostituzione
K 5 -2 -1 - 7 3 6 Nonostante K e R siano due amminoacidi diversi , hanno uno score positivo. Perchè? Sono entrambi amminoacidi carichi positivamente. AKRANR KAAANK -1 + (-1) + (-2) = 11
 + (-2) = 11.")
28
MATRICI PAM (Dayhoff et al. 1978) Sono basate sul concetto di mutazione puntiforme accettata, Point Accepted Mutation (PAM). Le prime matrici PAM sono state compilate in base all’analisi delle sostituzioni osservate in un dataset costituito da diversi gruppi di proteine omologhe, ed in particolare su 1572 sostituzioni osservate in 71 gruppi di sequenze di proteine omologhe con similarita’ molto alta (85% di identita’). La scelta di proteine molto simili era motivata dalla semplicita’ dell’allineamento, senza necessita’ di introdurre correzioni per le multiple hits (sostituzioni come A->G->A or A->G->N). L’analisi degli allineamenti mostro’ come diverse sostituzioni aminoacidiche si presentassero con frequenze anche molto differenti: le sostituzioni che non alterano seriamente la funzione della proteina, quelle “accettate” dalla selezione, si osservano piu’ di frequente di quelle distruttive.
. La scelta di proteine molto simili era motivata dalla semplicita’ dell’allineamento, senza necessita’ di introdurre correzioni per le multiple hits (sostituzioni come A->G->A or A->G->N). L’analisi degli allineamenti mostro’ come diverse sostituzioni aminoacidiche si presentassero con frequenze anche molto differenti: le sostituzioni che non alterano seriamente la funzione della proteina, quelle accettate dalla selezione, si osservano piu’ di frequente di quelle distruttive.")
29
(Henikoff and Henikoff, 1992)
MATRICI PAM La frequenza osservata per ciascuna specifica sostituzione (es. A G) puo’ essere usata per stimare la probabilita’ della transizione corrispondente in un allineamento di proteine omologhe. Le probabilita’ di tutte le possibili sostituzioni sono riportate nella matrice PAM Matrici BLOSUM (Henikoff and Henikoff, 1992) Matrici di sostituzione derivate dall’analisi di oltre 2000 blocchi di allineamenti multipli di sequenze, che riguardavano regioni conservate di sequenze correlate. Per ridurre il contributo di coppie di amminoacidi di proteine altamente correlate, gruppi di sequenze molto simili sono state trattate come se fossero sequenze singole ed e’ stato calcolato il contributo medio di ciascuna posizione. Utilizzando diversi cut-off per il raggruppamento di sequenze simili si sono ottenute diverse matrici BLOSUM. BLOSUM62, BLOSUM80, …
 puo’ essere usata per stimare la probabilita’ della transizione corrispondente in un allineamento di proteine omologhe. Le probabilita’ di tutte le possibili sostituzioni sono riportate nella matrice PAM. Matrici BLOSUM. (Henikoff and Henikoff, 1992) Matrici di sostituzione derivate dall’analisi di oltre 2000 blocchi di allineamenti multipli di sequenze, che riguardavano regioni conservate di sequenze correlate. Per ridurre il contributo di coppie di amminoacidi di proteine altamente correlate, gruppi di sequenze molto simili sono state trattate come se fossero sequenze singole ed e’ stato calcolato il contributo medio di ciascuna posizione. Utilizzando diversi cut-off per il raggruppamento di sequenze simili si sono ottenute diverse matrici BLOSUM. BLOSUM62, BLOSUM80, …")
30
(Henikoff and Henikoff, 1992)
MATRICI BLOSUM (Henikoff and Henikoff, 1992) Blocks Substitution Matrix Score derivati da frequenze di sostituzioni in blocchi di allineamenti locali in proteine correlate Il nome della matricindica la distanza evolutiva BLOSUM62 è stata creata usando sequenze che non avevano più del 62% di identità
 Blocks Substitution Matrix. Score derivati da frequenze di sostituzioni in blocchi di allineamenti locali in proteine correlate. Il nome della matricindica la distanza evolutiva. BLOSUM62 è stata creata usando sequenze che non avevano più del 62% di identità.")
31
PAMX PAMx = PAM1x PAM250 = PAM1250 PAM250 è una matrice molto usata:
Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys ... A R N D C Q E G H I L K ... Ala A Arg R Asn N Asp D Cys C Gln Q ... Trp W Tyr Y Val V

32
BLOSUM 80 A R N D C Q E G H I L K M F P S T W Y V B Z X * A 7 R -3 9
*

33
L’utilizzo della matrice di similarita’ appropriata per ciascuna analisi e’ cruciale per avere buoni risultati. Infatti relazioni importanti da un punto di vista biologico possono essere indicate da una significativita’ statistica anche molto debole. Sequenze poco divergenti molto divergenti BLOSUM80 BLOSUM62 BLOSUM45 PAM1 PAM120 PAM250

34
Il punteggio complessivo risultera’:
CALCOLO DEL PUNTEGGIO PER UN ALLINEAMENTO Data una coppia di sequenze Sa e Sb Per ogni coppia di elementi ai e bj di Sa e Sb si definisce un punteggio s(ai,bj) s(ai,bj) = se ai = bj s(ai,bj) = se ai bj , con > Ad ogni ogni gap viene assegnato un punteggio dato da: Wk = + (k-1) Dove Wk e’ una funzione lineare che assegna una penalita’ constante alla presenza del gap (, ad es. -10) e una penalita’ proporzionale alla lunghezza del gap meno uno. (gap opening penalty, GOP) (gap extension penalty, GEP) Il punteggio complessivo risultera’: (s(ai,bj) ) + (Wk) SIMILARITY SCORE MISMATCHES MATCHES GAP PENALTY GAPS
 s(ai,bj) = se ai = bj. s(ai,bj) = se ai bj , con > Ad ogni ogni gap viene assegnato un punteggio dato da: Wk = + (k-1) Dove Wk e’ una funzione lineare che assegna una penalita’ constante alla presenza del gap (, ad es. -10) e una penalita’ proporzionale alla lunghezza del gap meno uno. (gap opening penalty, GOP) (gap extension penalty, GEP) Il punteggio complessivo risultera’: (s(ai,bj) ) + (Wk) SIMILARITY SCORE. MISMATCHES. MATCHES. GAP PENALTY. GAPS.")
35
CALCOLO DEL PUNTEGGIO PER UN ALLINEAMENTO
Sequenze: Possibile allineamento: ATTCCGAG AGAC Assegno i seguenti punteggi: Match: +2 Mismatch: -1 GOP: -5 GEP: -2 MATCHES 3 3 x 2 = 6 MISMATCHES 1 1 x –1 = -1 SIMILARITY SCORE 6 –1 = 5 GAPS 1 (lungo 4 nucleotidi) GOP + GEP X 3 GOP -5 GEP -2 x 3 GAP PENALTY (3 x –2) = -11 PUNTEGGIO FINALE 5 – 11 = -6 ATTCCGAG | || A----GAC
 GOP + GEP X 3. GOP -5. GEP -2 x 3. GAP PENALTY -5 + (3 x –2) = -11. PUNTEGGIO FINALE 5 – 11 = -6. ATTCCGAG. | || A----GAC.")
36
ALGORITMI PER L’ALLINEAMENTO DI SEQUENZE Algoritmo di Needleman & Wunsch allineamento globale Algoritmo di Smith & Waterman allineamento locale

37
Manhattan Tourist Problem (MTP)
Siamo a manhattan! Abbiamo molte cose da visitare e solo strade a senso unico. Vogliamo determinare il percorso che ci porta da un estremo all’altro del quartiere e che ci premette di visitare il massimo numero di attrazioni

38
Manhattan Tourist Problem (MTP)
Source Imagine seeking a path (from source to sink) to travel (only eastward and southward) with the most number of attractions (*) in the Manhattan grid * * * * * * * * * * * Sink
 to travel (only eastward and southward) with the most number of attractions (*) in the Manhattan grid. * * * * * * * * * * * Sink.")
39
MTP: An Example source sink 3 5 9 13 15 19 20 23 j coordinates
1 2 3 4 j coordinates source 3 2 4 3 5 9 1 4 3 2 3 2 4 2 13 1 1 4 6 5 2 7 3 4 15 19 i coordinates 2 4 4 5 2 1 3 3 2 3 20 3 5 6 8 5 1 3 2 2 sink 23 4

40
MTP: Greedy Algorithm Is Not Optimal
1 2 5 source 5 3 10 5 2 1 5 3 5 3 1 2 3 4 promising start, but leads to bad choices! 5 2 22 sink 18

41
MTP: An Example source sink 3 5 9 9 1 4 7 13 15 17 5 10 20 24 9 14 22
1 2 3 4 j coordinates source 3 2 4 3 5 9 9 1 4 3 2 1 3 4 2 7 4 2 13 15 1 1 4 6 5 2 7 17 3 4 5 10 20 24 i coordinates 2 4 4 5 2 1 3 3 2 3 9 14 22 22 25 3 5 6 8 5 14 1 20 3 30 2 32 2 sink 35 4

42
ALGORITMO DI NEEDLEMAN & WUNSCH PER L’ALLINEAMENTO GLOBALE
Questo metodo permette di determinare l’allineamento globale ottimale attraverso un’interpretazione computazionale della matrice dotplot. L’idea e’ di calcolare ricorsivamente l’allineamento ottimo per sottosequenze via via piu’ lunghe, cosa possibile in virtu’ dell’indipendenza e dell’additivita’ dei punteggi. Le sequenze vengono comparate attraverso una matrice 2D, le celle rappresentanti matches hanno punteggio 1; 0 per i mismatches. L’algoritmo prevede una serie di somme successive dei punteggi contenuti nelle celle, che da’ luogo ad una matrice di punteggi, la cui analisi permette la costruzione dell’allineamento. Iniziando dalla casella piu’ in basso e piu’ a destra ( M(y,z) ), il valore massimo contenuto nella caselle della riga y e della colonna z viene sommato a quello nelle caselle della linea i=y-1 e della colonna j=z-1. Alla fine delle iterazioni il punteggio della cella piu’ in alto a sinistra rappresenta il punteggio totale dell’allineamento, senza considerare le gap penalties.
 ), il valore massimo contenuto nella caselle della riga y e della colonna z viene sommato a quello nelle caselle della linea i=y-1 e della colonna j=z-1. Alla fine delle iterazioni il punteggio della cella piu’ in alto a sinistra rappresenta il punteggio totale dell’allineamento, senza considerare le gap penalties.")
43
1 1 2 2 3 4 3 5 4 5 6 6 7 7 8 i coords: elements of v A T C T G A T C
1 1 2 2 3 4 3 5 4 5 6 6 7 7 8 i coords: elements of v A T -- C -- T G A T C elements of w -- T G C A T -- A -- C j coords: (0,0) (1,0) (2,1) (2,2) (3,3) (3,4) (4,5) (5,5) (6,6) (7,6) (8,7)
 (1,0) (2,1) (2,2) (3,3) (3,4) (4,5) (5,5) (6,6) (7,6) (8,7)")
45
Needleman-Wunsch FASE 1 R F C W T Y P D W K F W T P D Y A Identificazione residui identici Needleman-Wunsch FASE 2 R F C W T Y P D W K F W T P D P Y D A Somme successive

46
RFCWTYPD----WK -F-WT-PDPYDAW- Needleman-Wunsch Needleman-Wunsch FASE 3
Tracciare i percorsi massimali Needleman-Wunsch FASE 4 RFCWTYPD----WK -F-WT-PDPYDAW- Deduzione allineamento ottimale

47
Allineamento locale. Perchè?
Un gene in due specie diverse possono presentare delle brevi regioni di similarità pur essendo diversi nelle restanti regioni Esempio: I geni Homeobox hanno una regione chiamata homeodomain che è altamente conservata tra le specie. Un all’ineamento globale non sarebbe in grado di individuare questi domini

48
ALGORITMO DI SMITH & WATERMAN PER L’ALLINEAMENTO LOCALE
Lo scopo degli algoritmi di allineamento locale di due sequenze e’ trovare la regioni piu’ lunga della prima sequenza che produce un allineamento ottimale, dati certi parametri, con una regione della seconda.

49
ALGORITMO DI SMITH & WATERMAN PER L’ALLINEAMENTO LOCALE
Lo scopo degli algoritmi di allineamento locale di due sequenze e’ trovare la regioni piu’ lunga della prima sequenza che produce un allineamento ottimale, dati certi parametri, con una regione della seconda. Per misurare la bonta’ degli allineamenti si definiscono due funzioni: SIMILARITY SCORE, dipende dal PUNTEGGIO PER IL MATCH di residui ad es. 2 match, -1 mismatch (oppure A con A +2, A con G +1, …) GAP PENALTY W=a+b(k-1) ) a = GOP gap opening penalty b = GEP gap extension penalty ES: ATTCCGAG match +2, mismatch –1, GOP -5, GEP -2 | || A----GAC MATCHES 3 x 2 = 6 MISMATCHES 1 x –1 = -1 GAPS 1 lungo 4 nucleotidi (3 x –2) Anche il metodo di Smith and Waterman utilizza una matrice per comparare le due sequenze, in cui il valore numerico contenuto in ciascuna cella rappresenta il punteggio dell’allineamento locale ha inizio dai due residui corrispondenti. Cosi’, l’identificazione dei punteggi piu’ alti nella matrice permette di trovare i migliori allineamenti locali tra le due sequenze.
 GAP PENALTY W=a+b(k-1) ) a = GOP gap opening penalty. b = GEP gap extension penalty. ES: ATTCCGAG match +2, mismatch –1, GOP -5, GEP -2. | || A----GAC. MATCHES 3 x 2 = 6. MISMATCHES 1 x –1 = -1. GAPS 1 lungo 4 nucleotidi -5 + (3 x –2) Anche il metodo di Smith and Waterman utilizza una matrice per comparare le due sequenze, in cui il valore numerico contenuto in ciascuna cella rappresenta il punteggio dell’allineamento locale ha inizio dai due residui corrispondenti. Cosi’, l’identificazione dei punteggi piu’ alti nella matrice permette di trovare i migliori allineamenti locali tra le due sequenze.")
50
ALLINEAMENTO LOCALE: Free Rides
Yeah, a free ride! Origine (0,0) Le linee tratteggiate rappresentano I free rides che partono dall’origine (0,0) verso tutti gli altri
 Le linee tratteggiate rappresentano I free rides che partono dall’origine (0,0) verso tutti gli altri.")
51
RICERCA DI SIMILARITÀ Una sequenza “da sola” non e’ informativa, deve essere analizzata comparativamente al contenuto dei database perche’ possano essere formulate delle ipotesi sulla sue relazioni evolutive con sequenze simili o sulla sua funzione. Domande cui si puo’ rispondere con una ricerca di similarita’: Data una sequenza, ci sono cose simili nel database? Ho trovato un nuovo gene o una nuova proteina? Il gene ha somiglianze con qualche altro gene nella stessa specie o in altre specie? Fare ipotesi sulla funzione di una proteina Trovare le regione di sovrapposizione tra sequenze contigue Trovare la regione genomica codificante un trascritto Studiare l’evoluzione di popolazioni o specie

52
Basic Local Alignment Search Tool
BLAST Basic Local Alignment Search Tool (Altschul 1990) L’ algoritmo di BLAST e’ euristico e opera: Tagliando le sequenze da comparare in piccoli pezzi (parole) Ignorando tutte le coppie di parole (sequenza query/database) la cui comparazione da’ un punteggio inferiore ad un limite fissato Cercando di estendere tutte le hits rimanenti sino a che l’allineamento locale raggiunge un certo punteggio Dati una SEQUENZA QUERY ed un DATABASE DI SEQUENZE, BLAST ricerca nel database “parole” di lunghezza almeno “W” con un punteggio di similarita’ di almeno “T” una volta allineate con la sequenza “query” (HSP, High Scoring Pairs). Le “parole” selezionate vengono estese, se possibile, fino a raggiungere un punteggio superiore a “S” oppure un “E-value” inferiore al limite specificato.
 L’ algoritmo di BLAST e’ euristico e opera: Tagliando le sequenze da comparare in piccoli pezzi (parole) Ignorando tutte le coppie di parole (sequenza query/database) la cui comparazione da’ un punteggio inferiore ad un limite fissato. Cercando di estendere tutte le hits rimanenti sino a che l’allineamento locale raggiunge un certo punteggio. Dati una SEQUENZA QUERY ed un DATABASE DI SEQUENZE, BLAST ricerca nel database parole di lunghezza almeno W con un punteggio di similarita’ di almeno T una volta allineate con la sequenza query (HSP, High Scoring Pairs). Le parole selezionate vengono estese, se possibile, fino a raggiungere un punteggio superiore a S oppure un E-value inferiore al limite specificato.")
54
1- Seeding In sequenze di DNA W = 7 In sequenze proteiche W = 2-3

55
Two-hits algorithm Le word-hits tendono a clusterizzare lungo le diagonali L’algoritmo two-hits richiede che le word-hits siano entro una distanza prestabilita

56
2 - Extension La fase successiva comporta l’estensione dei seed
L’estensione avviene in entrambe le direzioni Blast ha un meccanismo per decidere quando fermare l’estensione

57
3 - Evaluation The quick brown fox jumps over the lazy dog.
The quiet brown cat purrs when she sees him. The quick brown fox jump The quiet brown cat purr <- score <- drop off score

58
La SIGNIFICATIVITA’ di un allineamento si calcola come P value o E value P value e’ la probabilita’ di ottenere un allineamento con punteggio uguale o migliore di quello osservato Si calcola mettendo in relazione il punteggio osservato (S) con la distribuzione attesa di HSP quando si comparano sequenze random della stessa lunghezza e composizione di quella in analisi (query sequence) Piu’ il Pvalue e’ vicino a 0 piu’ e’ significativo 2x e’ meglio do !!! E value e’ il numero atteso di allineamenti con punteggio uguale o migliore di quello osservato Piu’ e’ basso piu’ e’ buono

60
Usare BLAST OPZIONI Sequenza query nucleotidica proteica (sequenza in formato FASTA, GenBank Accession numbers o GI numbers) Database database di seq. nucleotidiche database di seq. proteiche Programma Standard BLAST (blastn) Standard protein BLAST (blastp) translated blast (blastx, tblastn, tblastx) MEGABLAST PSI-BLAST PHI-BLAST + altre opzioni … Blast selection table
 Standard protein BLAST (blastp) translated blast (blastx, tblastn, tblastx) MEGABLAST. PSI-BLAST. PHI-BLAST. + altre opzioni … Blast selection table.")
61
Usare BLAST database di seq. nucleotidiche
nr All GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or phase 0, 1 or 2 HTGS sequences). No longer "non-redundant". est Database of GenBank+EMBL+DDBJ sequences from EST division. est_human est_mouse htgs Unfinished High Throughput Genomic Sequences yeast Saccharomyces cerevisiae genomic nucleotide sequences mito Database of mitochondrial sequences vector Vector subset of GenBank(R), NCBI, in month All new or revised GenBank+EMBL+DDBJ+PDB sequences alu Select Alu repeats from REPBASE, suitable for masking Alu repeats from query sequences. dbsts Database of GenBank+EMBL+DDBJ sequences from STS division. chromosome Searches Complete Genomes, Complete Chromosome, or contigs form the NCBI Reference Sequence project.
. No longer non-redundant . est Database of GenBank+EMBL+DDBJ sequences from EST division. est_human. est_mouse. htgs Unfinished High Throughput Genomic Sequences. yeast Saccharomyces cerevisiae genomic nucleotide sequences. mito Database of mitochondrial sequences. vector Vector subset of GenBank(R), NCBI, in. month All new or revised GenBank+EMBL+DDBJ+PDB sequences. alu Select Alu repeats from REPBASE, suitable for masking Alu repeats from query sequences. dbsts Database of GenBank+EMBL+DDBJ sequences from STS division. chromosome Searches Complete Genomes, Complete Chromosome, or contigs form the NCBI Reference Sequence project.")
62
Usare BLAST PROGRAMMI Blastn Nucleotide query - Nucleotide db
Blastp Protein query - Protein db Translating BLAST attraverso la traduzione concettuale della query sequence o dei database permette di comparare una sequenza nucleotidica con database di proteine o viceversa. Translated query - Protein db blastx Protein query - Translated db tblastn Translated query - Translated db tblastx MEGABLAST usa un algoritmo greedy (ingordo) veloce ed ottimizzato per comparare sequenze che differiscono poco Search for short nearly exact matches blastn con parametri scelti in modo da ottimizzare la ricerca di matches quasi esatti e brevi. Questi si trovano spesso per caso, percio’ utilizza alto E-value, piccola dimensione della parola e filtering PSI-BLAST Find members of a protein family or build a custom position- specific score matrix PHI-BLAST Find proteins similar to the query around a given pattern
 veloce ed ottimizzato per comparare sequenze che differiscono poco. Search for short nearly exact matches blastn con parametri scelti in modo da ottimizzare la ricerca di matches quasi esatti e brevi. Questi si trovano spesso per caso, percio’ utilizza alto E-value, piccola dimensione della parola e filtering. PSI-BLAST Find members of a protein family or build a custom position- specific score matrix. PHI-BLAST Find proteins similar to the query around a given pattern.")
Presentazioni simili















 Algoritmi di allineamento 2) Algoritmi di ricerca in database>")




 BLAST: Altshul (1990)>")

