Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
MALATTIE METABOLICHE EREDITARIE
MAURIZIO SCARPA DIPARTIMENTO DI PEDIATRIA CENTRO REGIONALE MALATTIE RARE

2
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI
A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA Le malattie metaboliche sono patologie rare, con una frequenza complessiva di 1:5000 nati vivi, generalmente ereditate come malattie autosomiche recessive. La malattia metabolica più frequente la Fenilchetonuria con una frequenza di 1: Generalmente queste malattie esitano con gravi quadri di compromissione neurologica, ritardo mentale e morte. Fortunatamente, per alcune patologie, molti effetti gravi possono essere prevenuti con una diagnosi precoce ed un adeguato e tempestivo trattamento dietetico.

3
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI
A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA Le malattie metaboliche sono causate da difetti enzimatici che causano un alterato processamento di molecole semplici, aminoacidi, o complesse, carboidrati e lipidi. Il deficit enzimatico previene il metabolismo normale di un nutriente o di un suo metabolita fino a raggiungere livelli tossici. Inoltre, alcuni pazienti possono sviluppare delle carenze nutrizionali perché incapaci di produrre il prodotto metabolico finale.

4
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI
A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA L’anormale metabolismo causa effetti importanti nell’organismo. Carenze energetiche o nutrizionali comportano un blocco dello sviluppo dell’individuo con potenziali danni permanenti cerebrali in caso di raggiungimento di livelli tossici del metabolita alterato. Le alterazioni dovute ad accumulo tossico possono variare a seconda del tipo di molecola accumulata nell’unità di tempo.

5
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI
A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA Benchè gli effetti potenziali di un difetto metabolico possano essere estremamente gravi, gli stessi possono essere ridotti mediante un attento regime dietetico. Molti bambini con malattie metaboliche si sviluppano e vivono in maniera normale. Se non trattati o trattati inappropriatamente i piccoli pazienti sviluppano, nella maggior parte dei casi, ritardo mentale, deficit neurologico, coma e morte.

6
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI
A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA Il primo trattamento dietietico di una malattia metabolica è stato descritto nel 1954 dal Dr. Horst Bickel, il quale riportò i benefici neurologici in pazienti affetti da fenilchetonuria (PKU) dopo dieta senza fenilalanina. Sfortunatamente questi pazienti avevano già sviluppato un grave ritardo mentale. Dopo questa comunicazione, l’applicazione di una dieta adeguata in caso di PKU ha permesso di prevenire il ritardo mentale adottando una dieta a basso regime di fenilalanina già in epoca neonatale..
 dopo dieta senza fenilalanina. Sfortunatamente questi pazienti avevano già sviluppato un grave ritardo mentale. Dopo questa comunicazione, l’applicazione di una dieta adeguata in caso di PKU ha permesso di prevenire il ritardo mentale adottando una dieta a basso regime di fenilalanina già in epoca neonatale..")
8
Metabolic Disorders Metabolic Disorders Metabolic Disorders Metabolic Disorders Metabolic Disorders
Taken as a whole are common There are over 500 Defined and the number is growing As is our understanding and ability to diagnose and treat

9
Metabolic Disorders Diagnosis is difficult due to :
The nonspecific nature of clinical presentation (poor feeding, altered mental status, developmental delay) Lack of family history, being autosomal recessive Diagnosis is critical as early treatment significantly improves outcome for many.
 Lack of family history, being autosomal recessive. Diagnosis is critical as early treatment significantly improves outcome for many.")
10
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA
SINTOMI NON SPECIFICI IN UN CONTESTO CLINICO EVOCATIVO Neonato a termine da gravidanza normale Intervallo senza sintomi Deterioramento progressivo , senza causa apparente Esami di routine nella norma (compresi Rx torace, esame del liquor, indagini microbiologiche, neuroimaging) Il peggioramento clinico, inaspettato e inspiegabile di un neonato, dopo un iniziale periodo senza sintomi è il segnale più importante della presenza di una patologia metabolica ereditaria di tipo "intossicazione".
 Il peggioramento clinico, inaspettato e inspiegabile di un neonato, dopo un iniziale periodo senza sintomi è il segnale più importante della presenza di una patologia metabolica ereditaria di tipo intossicazione .")
11
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA
PRIMO IMPATTO: SINTOMI NON SPECIFICI Problemi respiratori, distress respiratorio Ipotonia Suzione scarsa Vomito Disidratazione Letargia Convulsioni FONDAMENTALE: Attivare Indagini eziologiche rivolte a documentare le cause più frequenti (infezioni) VALORIZZARE: Consanguineità Precedenti decessi in epoca neonatale, di nascita a termine di gravidanza regolare, di intervallo libero dalla nascita alla comparsa dei sintomi
 VALORIZZARE: Consanguineità. Precedenti decessi in epoca neonatale, di nascita a termine di gravidanza regolare, di intervallo libero dalla nascita alla comparsa dei sintomi.")
12
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA
Rivalutazione del neonato - Il sospetto clinico è cruciale per l'identificazione del neonato a rischio di patologia metabolica ereditaria. - I segni e i sintomi interpretati come esito di ipossia , infezione o altre cause comuni diventano evocativi soprattutto in assenza di chiare cause eziologiche. Comune alla maggior parte degli errori congeniti del metabolismo ad esordio neonatale è la sintomatologia neurologica (alterazioni del tono muscolare, della reattività, convulsioni) ATTENZIONE: l'encefalopatia metabolica può accompagnarsi a sintomi respiratori (alcalosi o acidosi respiratoria), apnea, bradicardia e ipotermia. Spesso è presente ipocalcemia, ipo- iper glicemia. A volte leucopenia e piastrinopenia. L'errore diagnostico più comune è porre diagnosi di sepsi.
 ATTENZIONE: l encefalopatia metabolica può accompagnarsi a sintomi respiratori (alcalosi o acidosi respiratoria), apnea, bradicardia e ipotermia. Spesso è presente ipocalcemia, ipo- iper glicemia. A volte leucopenia e piastrinopenia. L errore diagnostico più comune è porre diagnosi di sepsi.")
13
INBORN ERRORS OF METABOLISM
Can be roughly divided into two groups based on cellular localization and clinical presentation. Group 1-Predominantly Cytoplasm Catabolism of Amino Acids, Fatty Acids and Carbohydrates. Present acutely after a brief asymptotic period Presentation may include reduced level of consciousness, respiratory distress, hypertonia, seizures, order Group 2-Predominantly Organellar Lysosomal storage and mitochondrial Have a more chronic course Often do not present until late childhood or adult hood

18
IDENTIFICAZIONE DEI NEONATI E DEI LATTANTI
A RISCHIO DI PATOLOGIA METABOLICA EREDITARIA DIAGNOSI DIFFERENZIALE Cardiomiopatia restrittiva Insufficienza cardiaca congestizia Epatiti Edema polmonare Ipoglicemia Meningiti ed Encefaliti Stenosi pilorica REYE Syndrome Apnee SIDS Gastroenterite Infezioni Vie Urinarie e Sepsi Pielonefriti

19
Screening Early Diagnosis = Early treatment Children Newborns Urine
Blood Blood or urine on fllter paper (spots) Newborns Dried blood spots
 Newborns. Dried blood spots.")
20
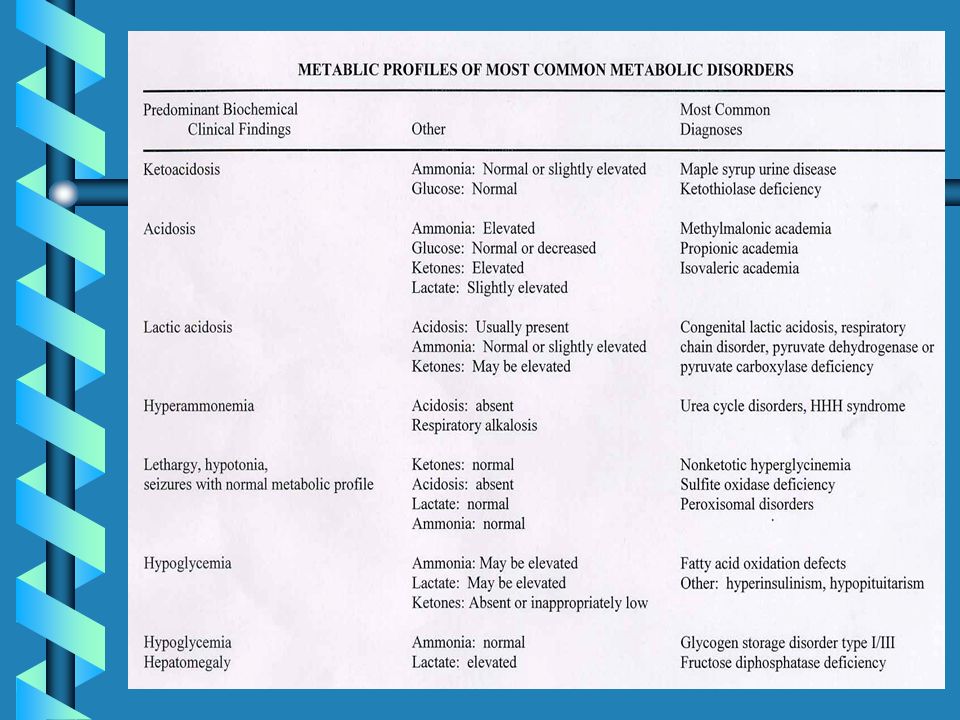
PATOLOGIE METABOLICHE EREDITARIE SCREENABILI MEDIANTE TANDEN MASSA
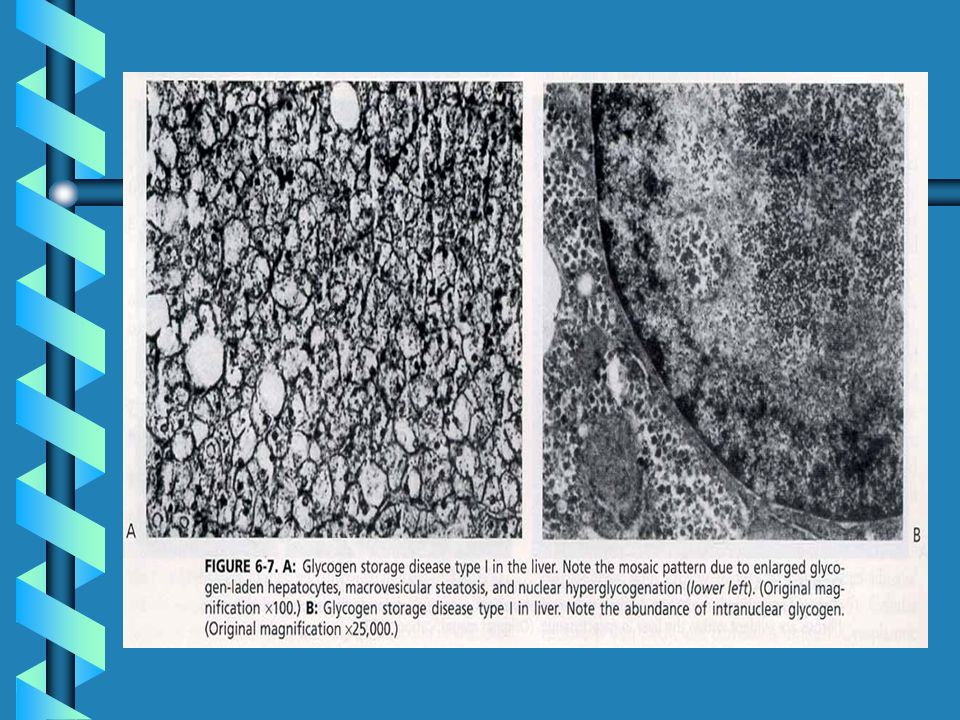
Difetti legati all'ossidazione degli acidi grassi Deficit di acil-CoA deidrogenasi per acidi grassi a corta catena (SCAD) Deficit di acil-CoA deidrogenasi per acidi grassi a media catena (MCAD) Deficit di acil-CoA deidrogenasi per acidi grassi a corta catena (LCAD) Deficit multiplo di acil-CoA deidrogenasi (MADD) Deficit di 3-OHacil-CoA deidrogenasi (LCHAD) Deficit di carnitina palmitoil transferasi (CPT-II) Deficit di 2,4-dienoil-CoA reduttasiDeficit di traslocasi Difetti legati al catabolismo degli amminoacidi ramificati Deficit di propionil-CoA carbossilasi (PA) Acidemia metilmalonica (tutti i tipi) (MMA) Deficit di isovaleril-CoA deidrogenasi (IVA) Deficit di metilcrotonil-CoA carbossilasi (3-MCC) Deficit di beta-chetotiolasi (beta-KT) Deficit di glutaril-CoA deidrogenasi (GA-I) Deficit di 3OH-3metilglutaril-CoA liasi (HMG)
 Deficit di acil-CoA deidrogenasi per acidi grassi a media catena (MCAD) Deficit di acil-CoA deidrogenasi per acidi grassi a corta catena (LCAD) Deficit multiplo di acil-CoA deidrogenasi (MADD) Deficit di 3-OHacil-CoA deidrogenasi (LCHAD) Deficit di carnitina palmitoil transferasi (CPT-II) Deficit di 2,4-dienoil-CoA reduttasiDeficit di traslocasi. Difetti legati al catabolismo degli amminoacidi ramificati Deficit di propionil-CoA carbossilasi (PA) Acidemia metilmalonica (tutti i tipi) (MMA) Deficit di isovaleril-CoA deidrogenasi (IVA) Deficit di metilcrotonil-CoA carbossilasi (3-MCC) Deficit di beta-chetotiolasi (beta-KT) Deficit di glutaril-CoA deidrogenasi (GA-I) Deficit di 3OH-3metilglutaril-CoA liasi (HMG)")
26
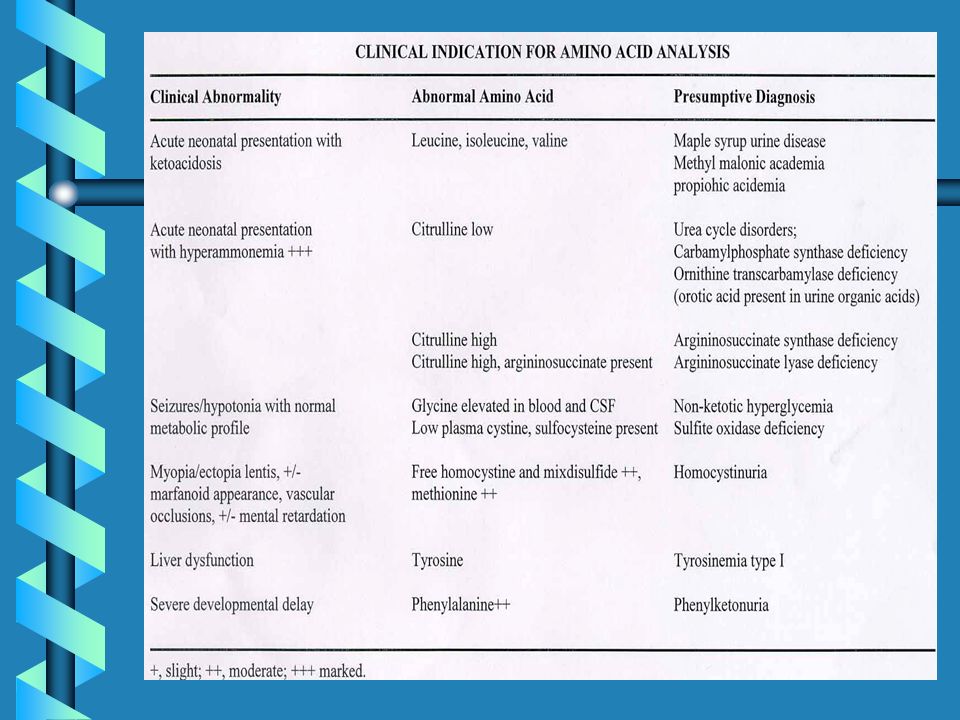
UREA CYCLE DISORDERS

27
UREA CYCLE DEFECTS Clinical Synopsis METABOLIC :
Episodic ammonia intoxication Respiratory alkalosis NEUROLOGIC : Irritability Lethargy Ataxia Coma Seizures Cerebral edema Developmental delay Mental retardation GASTROINTESTINAL : Protein avoidance Vomiting GROWTH : Failure to thrive DIAGNOSTIC LABORATORY : Hyperammonemia High plasma citrulline ( micromolar) High plasma glutamine Hepatic argininosuccinate synthetase deficiency MOLECULAR : Mutations in argininosuccinate synthetase INHERITANCE : Autosomal recessive
 High plasma glutamine. Hepatic argininosuccinate synthetase deficiency. MOLECULAR : Mutations in argininosuccinate synthetase. INHERITANCE : Autosomal recessive.")
28
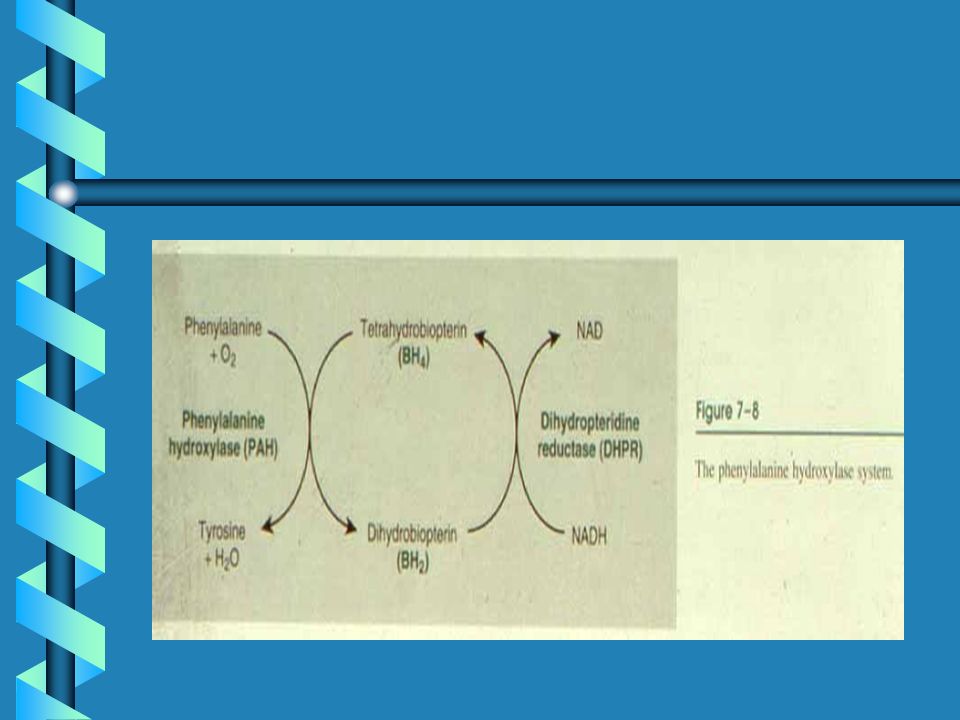
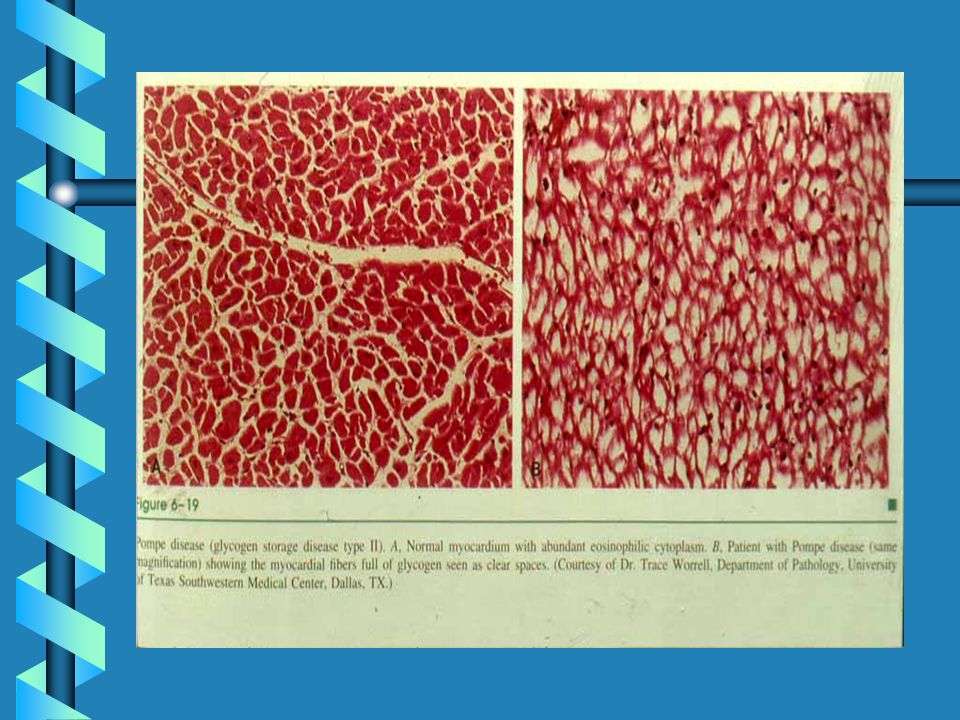
MAPLE SYRUP URINE DISEASE

29
MAPLE SYRUP URINE DISEASE
Clinical Synopsis INHERITANCE : Autosomal recessive GROWTH : Other Feeding problems ABDOMEN : Pancreas Pancreatitis Gastrointestinal Vomiting NEUROLOGIC : Central nervous system, Letharg,Seizures, Coma Mental retardation Hypertonia, Hypotonia, Cerebral edema METABOLIC FEATURES : Ketosis, Hypoglycemia, Lactic acidosis in E3-deficiency LABORATORY ABNORMALITIES : Elevated plasma branched chain amino acids (leucine, isoleucine, valine) Maple syrup urine odor Branched chain ketoaciduria (alpha-keto isocaproate, alpha-keto-beta methylisovalerate, alpha-keto isovalerate) Elevated plasma alloisoleucine Positive urine DNPH screening test MISCELLANEOUS : Five clinical variants of MSUD: (1) Classic severe form, (2) Intermittent form, (3) Intermediate form, (4) Thiamine-responsive form, (5) Dihydrolipoyl dehydrogenase (E3)-deficient In inbred Old Order Mennonite population of Lancaster County, MSUD prevalence is 1/176 newborns Onset of symptoms 4-7 days of age in classical severe form Death in untreated children MOLECULAR BASIS : Caused by mutations in the catalytic subunit genes of the branched-chain alpha-keto acid dehydrogenase complex, including E1 alpha subunit gene, E1 beta subunit gene E2 subunit gene E3 subunit gene
 Maple syrup urine odor. Branched chain ketoaciduria (alpha-keto isocaproate, alpha-keto-beta methylisovalerate, alpha-keto isovalerate) Elevated plasma alloisoleucine. Positive urine DNPH screening test. MISCELLANEOUS : Five clinical variants of MSUD: (1) Classic severe form, (2) Intermittent form, (3) Intermediate form, (4) Thiamine-responsive form, (5) Dihydrolipoyl dehydrogenase (E3)-deficient. In inbred Old Order Mennonite population of Lancaster County, MSUD prevalence is 1/176 newborns. Onset of symptoms 4-7 days of age in classical severe form. Death in untreated children. MOLECULAR BASIS : Caused by mutations in the catalytic subunit genes of the branched-chain alpha-keto acid dehydrogenase complex, including. E1 alpha subunit gene, E1 beta subunit gene E2 subunit gene E3 subunit gene.")
42
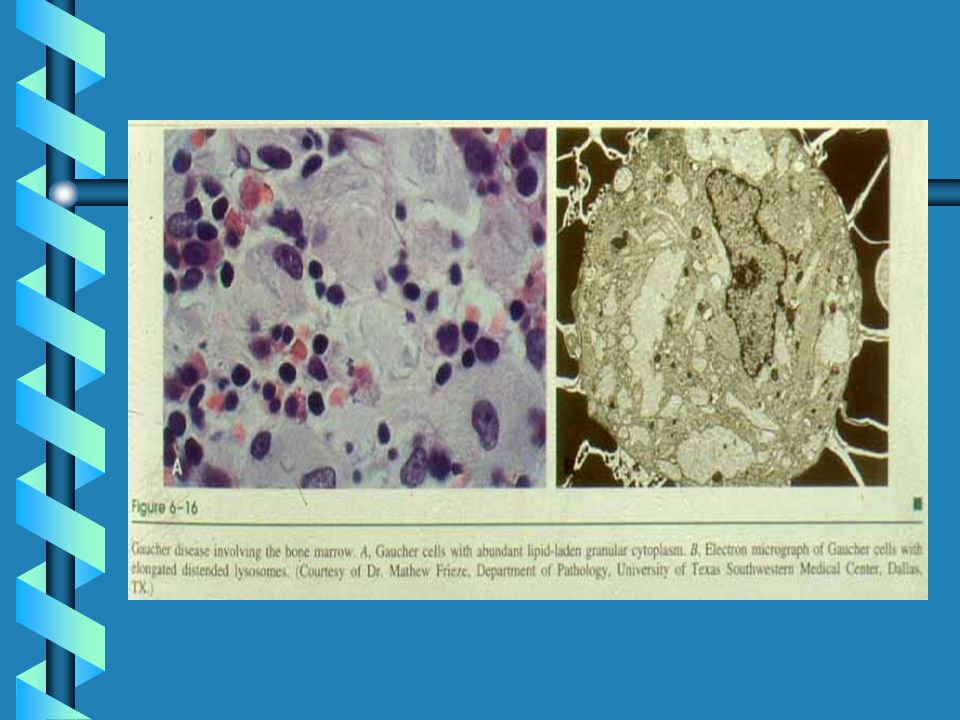
Glucocerebroside-laden Gaucher Cells

43
Gaucher Disease Type 1 Young Severe
Initially, patient had severe liver and spleen involvement Patient shown post-splenectomy. Liver fills abdominal cavity. Note bowing of legs due to long bone involvement. Rapid progressive disease signs are more commonly seen in children.

44
Gaucher Disease Type 2 Visceromegaly Strabismus Cortical thumbs
Retroflexion of the neck Failure to thrive Cachexia

45
Gaucher Disease Type 3 Typical presentation of severe early onset
Massive visceral enlargement Progressive developmental delay

46
Skeletal Complications of Gaucher Disease

47
Skeletal Pathology of Gaucher Disease
Haemorrhagic infarction Necrosis Osteosclerosis Severe osteoporosis Loss of cortical bone

48
Gaucher Cells in Bone Marrow
T1-weighted MR Image

49
Erlenmeyer Flask Deformity

50
Mineral Bone Damage

51
Platyspondyly

52
Thickened Interlobular Septa

53
T1 MRI of Splenic Nodules in Gaucher’s

54
Hepatosplenomegaly in Gaucher’s

55
Femur Before and After 48 Months of Ceredase Treatment

56
Gaucher Disease Before and After 3 1/2 Years of Ceredase Treatment

57
Gaucher Disease Before and After 33 Months of Ceredase Treatment

60
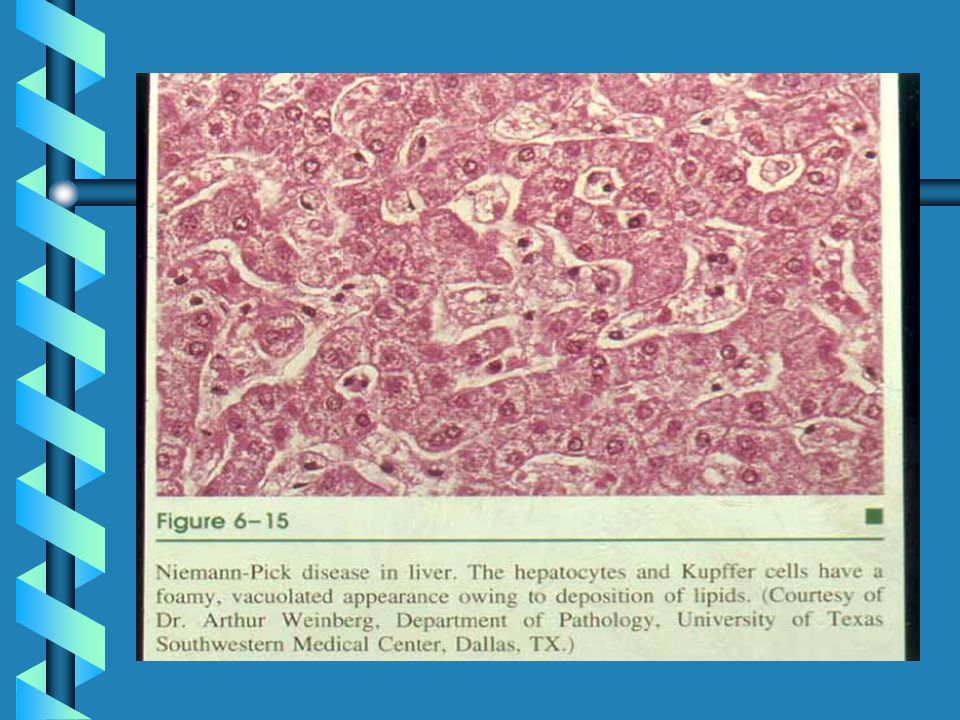
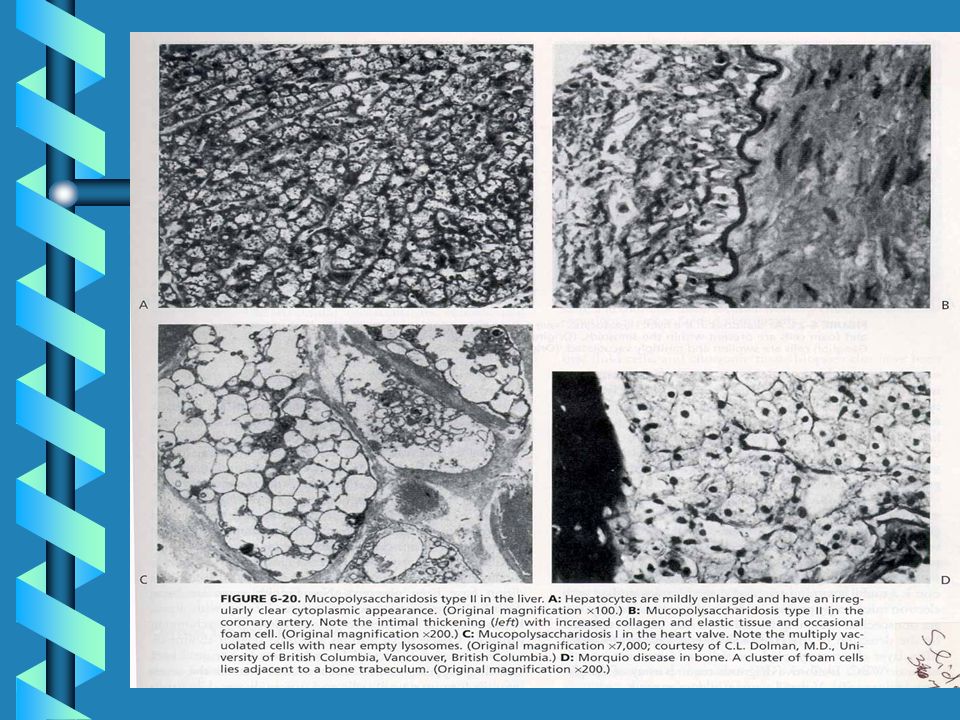
MUCOPOLYSACCHARIDOSES

62
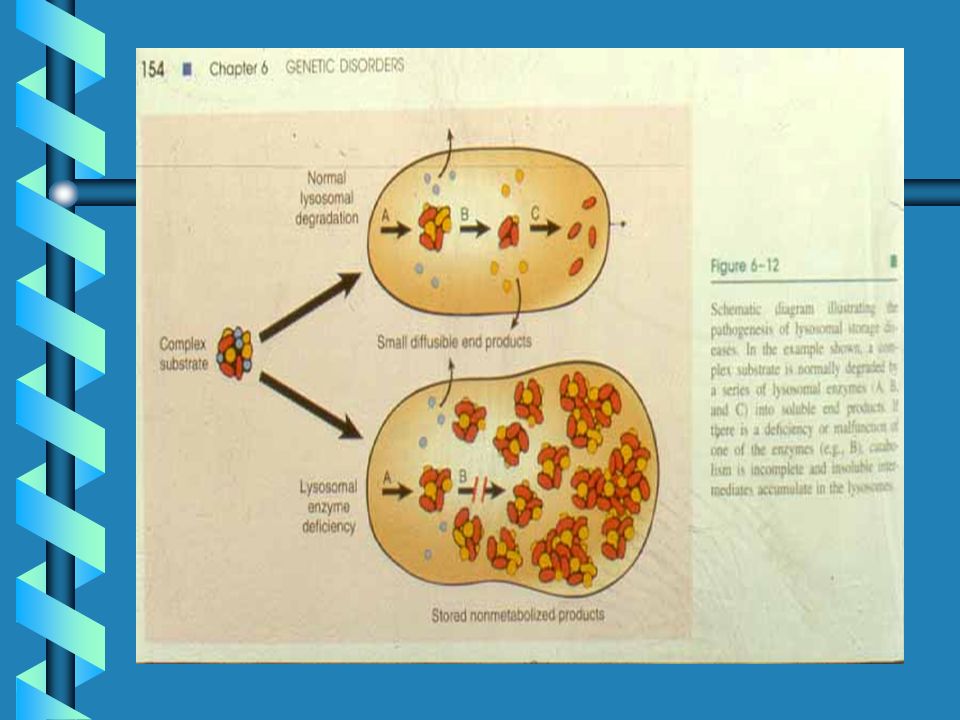
LSD ARE DUE TO GAG STORAGE

64
Lysosomal Storage of GAG
These light micrographs show GAG accumulation in the lysosomes of cells in severe MPS I. The image on the left is from the liver, showing distended lysosomes in hepatocytes and Kupffer cells. The image on the right is from a conjunctival biopsy, showing fibroblasts with multiple distended lysosomes. Arrows point to the general areas of GAG storage. (Buchino et al., 1997) Photo reproduced by permission of Arnold Publishers.
 Photo reproduced by permission of Arnold Publishers.")
65
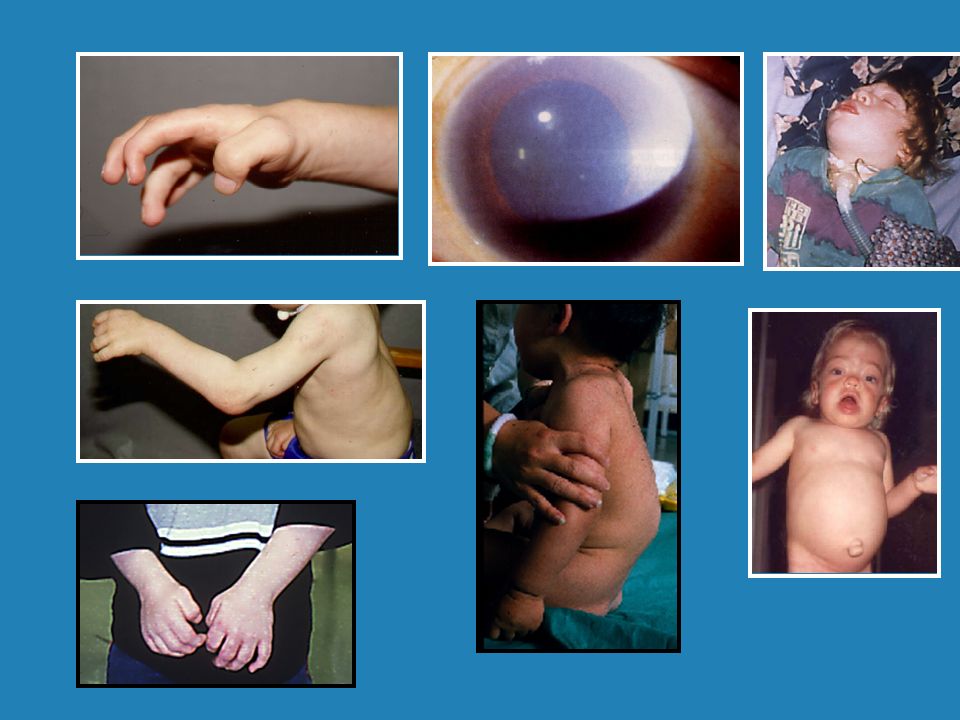
Multisystemic Manifestations
Neurologic Skeletal Respiratory Auditory Ocular Cardiovascular Gastrointestinal Progressive accumulation of GAG causes increasing involvement of multiple organ systems. Neurologic, skeletal, respiratory, ocular, cardiovascular, and gastrointestinal involvement are common complications of severe MPS I.

68
Disease Progression - Severe MPS I
12 months 10 months These photos help demonstrate the progressive nature of MPS I. This patient has the severe form of MPS I. 22 months 34 months 39 months

69
Disease Progression - Attenuated MPS I
3 years 4 years 6 years 8 years 11 years On the other end of the disease spectrum, this patient has the mild form of MPS I.

72
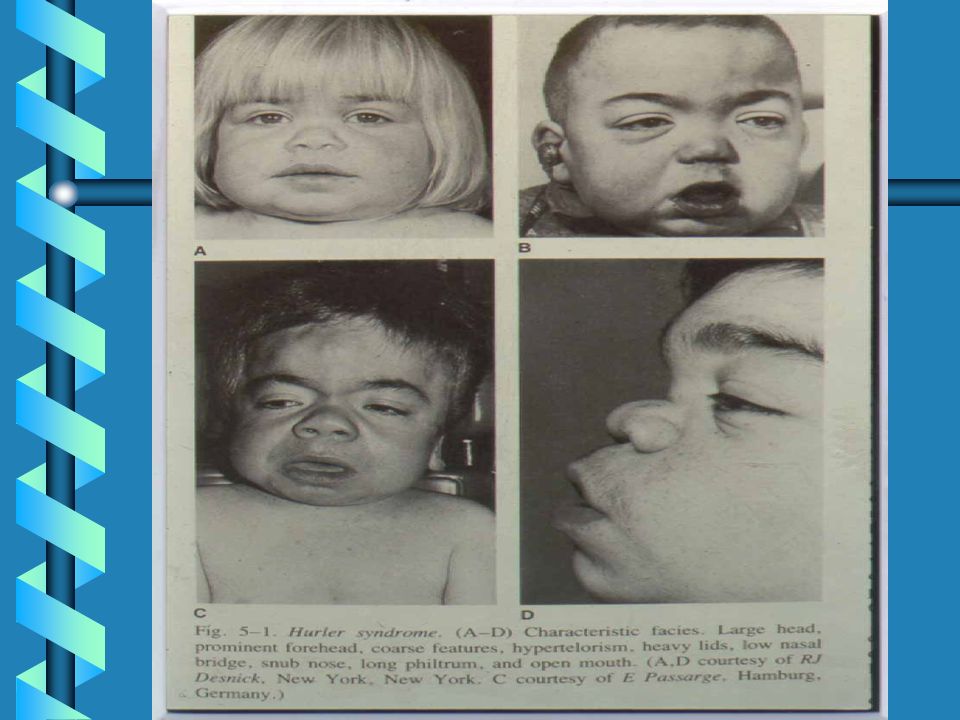
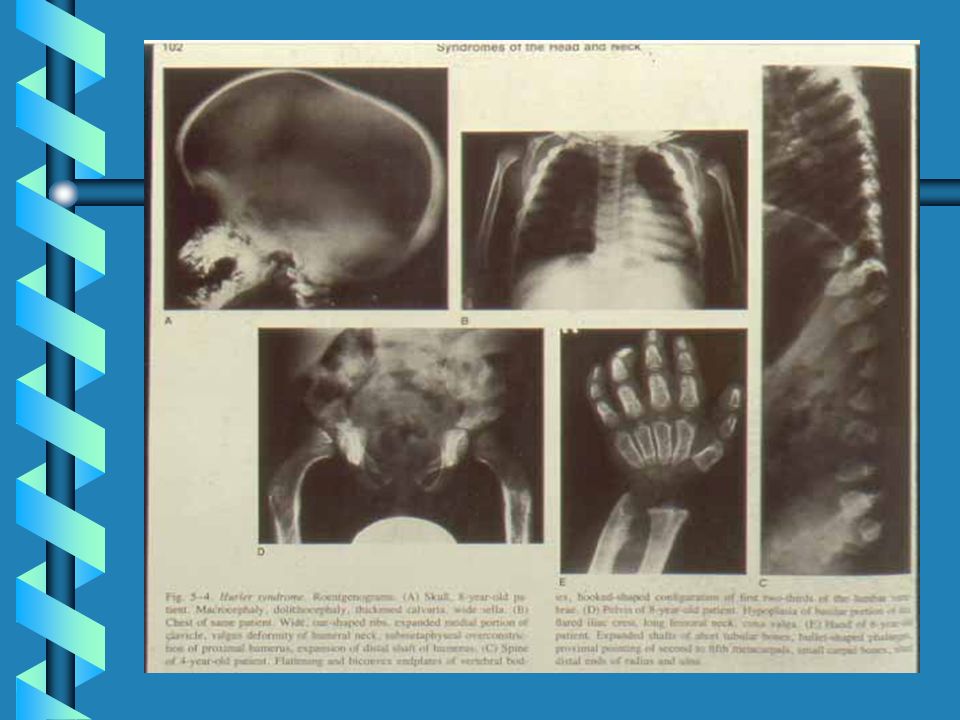
LE MUCOPOLISACCARIDOSI
MPS IHS Hurler-Scheie MPS IH Hurler MPS IS Scheie MPS II Hunter MPS II Hunter MPS IIIA Sanfilippo MPS IIIB Sanfilippo LE MUCOPOLISACCARIDOSI MPS IV Morquio MPS IV Morquio MPS VI Maroteaux-Lamy MPS VII Sly

73
Current Disease Management
Symptomatic management supportive care treatment of complications can improve quality of life Bone marrow transplantation more severe cases Enzyme Replacement Therapy Today, the management of MPS I patients consists of symptomatic management through supportive care and treatment of complications. Generally speaking, supportive or symptomatic management – with particular attention to the respiratory and cardiovascular complications, skeletal manifestations, arthropathy, loss of hearing and vision, gastrointestinal symptoms, and communicating hydrocephalus – can greatly improve the quality of life for patients and their families. For the most severe patients, bone marrow transplantation may be an option.

74
Clinical Studies Phase 1/2 Study (N=10) Phase 3 Study (N=45)
Phase 3 Extension Study (N=45) Compassionate Use Program (N=18)
 Compassionate Use Program (N=18)")
75
Phase 3 Study: Aldurazyme Reduced Hepatomegaly
Double-blind 26 wk Open-label Extension 24 wk 50 wk 5 1.3 Placebo/ Aldurazyme Aldurazyme/ -5 Mean % Change in Liver Volume -10 -12.6 We found that Aldurazyme effciant cleared GAG from the liver as evidenced by the reduction in hepatomegaly. In the Phase 3 DB study, patients receiving Aldurazyme showed a 18.9 % reduction in liver volume compared to a 1.3% increase in placebo patients after 26 weeks. The difference between groups was highly significant. Patients then rolled over into an open-label extension study where all patients received Aldurazyme. The placebo cross-ver patients now showed a 12.6% reduction in liver volume, and patients who received Aldurazyme throughout, there was an additional small reduction so these patients maintained their reduction in liver volume. Of patients with abnormal liver volumes, 64% (13+5/18+10) normalized after 6 months and 80% 12/15) normalized after 52 weeks. This shows that the dose is effective in clearing GAG substrate from the liver. The mean reduction and proportion of patients normalizing their liver volumes are consistent with the Phase 1/2 results. Clinical benefit? -15 -20 -18.9* -21.7 -25 *P=0.001 compared to placebo. Shift from abnormal to normal liver volume with Aldurazyme: 64% after 6 months, 80% after 12 months.
 normalized after 6 months and 80% 12/15) normalized after 52 weeks. This shows that the dose is effective in clearing GAG substrate from the liver. The mean reduction and proportion of patients normalizing their liver volumes are consistent with the Phase 1/2 results. Clinical benefit * *P=0.001 compared to placebo. Shift from abnormal to normal liver volume with Aldurazyme: 64% after 6 months, 80% after 12 months.")
76
Phase 3 Study: Aldurazyme Reduced Urinary GAG Levels
Double-blind Open-label Extension 300 Placebo patients begin Aldurazyme Placebo/Aldurazyme Aldurazyme/Aldurazyme -69% -65% +47% Mean Urinary GAG Levels (μg/mg Creatinine) P<0.001 150 Aldurazyme also greatly reduced urinary GAG levels. In the Phase 3 double-blind study, patients receiving Aldurazyme had a rapid decrease by the first timepoint at 4 weeks, and it remained low. Conversely, in placebo patients GAG levels showed an increase. The difference between groups, 101%, was highly significant. In the open-label extension study, when placebo patients now received Aldurazyme, the GAG levels dropped quickly and reached a level similar to that of patients who received Aldurazyme throughout. X patients had normal GAG levels by Week 50. The mean levels were approximately twice the upper limit of normal as indicated by the X color line. This is the upper limit of normal for adolescents, which corresponds to the mean age of the study population. The levels continue to come down slowly as seen with long-term treatment in the Phase 1/2 study. These pharmacodynamic studies confirm the Phase 1/2 results and indicate the the Aldurazyme dosing regimen rapdly and significantly clears GAG from the body and the decreased is sustained with long-term treatment. -54% Upper limit of normal for 13–18 yo age range (38.5 μg/mg creatinine) Baseline Week Week 50
 P< Aldurazyme also greatly reduced urinary GAG levels. In the Phase 3 double-blind study, patients receiving Aldurazyme had a rapid decrease by the first timepoint at 4 weeks, and it remained low. Conversely, in placebo patients GAG levels showed an increase. The difference between groups, 101%, was highly significant. In the open-label extension study, when placebo patients now received Aldurazyme, the GAG levels dropped quickly and reached a level similar to that of patients who received Aldurazyme throughout. X patients had normal GAG levels by Week 50. The mean levels were approximately twice the upper limit of normal as indicated by the X color line. This is the upper limit of normal for adolescents, which corresponds to the mean age of the study population. The levels continue to come down slowly as seen with long-term treatment in the Phase 1/2 study. These pharmacodynamic studies confirm the Phase 1/2 results and indicate the the Aldurazyme dosing regimen rapdly and significantly clears GAG from the body and the decreased is sustained with long-term treatment. -54% Upper limit of normal for 13–18 yo age range (38.5 μg/mg creatinine) Baseline Week 26 Week 50.")
77
Phase 3 Study: Aldurazyme Improved FVC Levels
Double-blind Open-label Extension 6 Placebo/Aldurazyme P=0.001* P=0.065† Aldurazyme/Aldurazyme 4 2 Mean Change in FVC, Percentage Points P=0.009* (between group) In the double-blind phase, patients receiving aldurazyme showed a mean increase of 4.6 percentage points at week 26, compared to a -0.7 mean decrease in placebo patients. The difference between groups was 5.3 percentage points and the difference between groups was significant. The changes were not smooth. We believe that the interim dip that occurred in both groups was due to a seasonal effect (this was springtime). At each timepoint, the Aldurazyme group showed a higher mean change than the placebo patients. The relative change from a baseline of 48% predicted was 10%. In the open-label extension, patients crossing over form placebo to aldurazyme showed an initial slight decline through the first 12 weeks, but then showed an increase over the last 24 weeks. The change from initiation of treatment was 2.6 percentage points, which approached statistical significance. Meanwhile, patients who receivied aldurazyme for 50 weeks maintained their improvement. and this change from baseline was significant. -2 Placebo patients begin Aldurazyme -4 Baseline Week Week 62 * Change from Baseline † Change from Week 26
 In the double-blind phase, patients receiving aldurazyme showed a mean increase of 4.6 percentage points at week 26, compared to a -0.7 mean decrease in placebo patients. The difference between groups was 5.3 percentage points and the difference between groups was significant. The changes were not smooth. We believe that the interim dip that occurred in both groups was due to a seasonal effect (this was springtime). At each timepoint, the Aldurazyme group showed a higher mean change than the placebo patients. The relative change from a baseline of 48% predicted was 10%. In the open-label extension, patients crossing over form placebo to aldurazyme showed an initial slight decline through the first 12 weeks, but then showed an increase over the last 24 weeks. The change from initiation of treatment was 2.6 percentage points, which approached statistical significance. Meanwhile, patients who receivied aldurazyme for 50 weeks maintained their improvement. and this change from baseline was significant. -2. Placebo patients. begin Aldurazyme. -4. Baseline Week 26 Week 62. * Change from Baseline † Change from Week 26.")
78
Phase 3 Study: Aldurazyme Increases 6MWT Distance
Double-blind Open-label Extension 50 Placebo/Aldurazyme 40 P=0.005* P=0.023† Aldurazyme/Aldurazyme 30 20 10 P=0.066* (between group) Mean Change, Meters -10 Aldurazyme also greatly reduced urinary GAG levels. In the Phase 3 double-blind study, patients receiving Aldurazyme had a rapid decrease by the first timepoint at 4 weeks, and it remained low. Conversely, in placebo patients GAG levels showed an increase. The difference between groups, 101%, was highly significant. In the open-label extension study, when placebo patients now received Aldurazyme, the GAG levels dropped quickly and reached a level similar to that of patients who received Aldurazyme throughout. X patients had normal GAG levels by Week 50. The mean levels were approximately twice the upper limit of normal as indicated by the X color line. This is the upper limit of normal for adolescents, which corresponds to the mean age of the study population. The levels continue to come down slowly as seen with long-term treatment in the Phase 1/2 study. These pharmacodynamic studies confirm the Phase 1/2 results and indicate the the Aldurazyme dosing regimen rapdly and significantly clears GAG from the body and the decreased is sustained with long-term treatment. -20 -30 Placebo patients begin Aldurazyme -40 -50 Baseline Week Week 62 *Change from Baseline. †Change from Week 26.
 Mean Change, Meters Aldurazyme also greatly reduced urinary GAG levels. In the Phase 3 double-blind study, patients receiving Aldurazyme had a rapid decrease by the first timepoint at 4 weeks, and it remained low. Conversely, in placebo patients GAG levels showed an increase. The difference between groups, 101%, was highly significant. In the open-label extension study, when placebo patients now received Aldurazyme, the GAG levels dropped quickly and reached a level similar to that of patients who received Aldurazyme throughout. X patients had normal GAG levels by Week 50. The mean levels were approximately twice the upper limit of normal as indicated by the X color line. This is the upper limit of normal for adolescents, which corresponds to the mean age of the study population. The levels continue to come down slowly as seen with long-term treatment in the Phase 1/2 study. These pharmacodynamic studies confirm the Phase 1/2 results and indicate the the Aldurazyme dosing regimen rapdly and significantly clears GAG from the body and the decreased is sustained with long-term treatment Placebo patients. begin Aldurazyme Baseline Week 26 Week 62. *Change from Baseline. †Change from Week 26.")
79
Phase 3 Study: Aldurazyme Increases Shoulder Flexion
Shoulder Flexion Median (90.5 o) at Baseline Double-blind 26 wk Open-label Extension 24 wk 50 wk 20 15.2 15 9.6 8.7 Placebo/ Aldurazyme Aldurazyme/ 10 Degrees Trends in other joint range of motion. Patients were not sleected for shoulder flexion, so patients with less restriction will limit the apparent treatment effect when considered as a mean group change. Similar to results seen in the Phase 1/2 study in shoulder flexion. 5 -5 -4.8 -10 n=12 n= n=12 n=9
 at Baseline. Double-blind. 26 wk. Open-label Extension. 24 wk 50 wk Placebo/ Aldurazyme. Aldurazyme/ 10. Degrees. Trends in other joint range of motion. Patients were not sleected for shoulder flexion, so patients with less restriction will limit the apparent treatment effect when considered as a mean group change. Similar to results seen in the Phase 1/2 study in shoulder flexion n=12 n=7 n=12 n=9.")
80
The End

Presentazioni simili

















 Brussels, 26 settembre 2013.>")






>")








>")


 17 Ottobre 2009 DOPPIA ANTIAGGREGAZIONE PIASTRINICA.>")