Scaricare la presentazione
1
FIBROSI CISTICA O MUCOVISCIDOSI
La Fibrosi Cistica (FC) o Mucoviscidosi è una grave malattia congenita, evolutiva e cronica, che colpisce indifferentemente maschi e femmine, trasmessa con meccanismo autosomico recessivo, il cui difetto di base consiste nella produzione di una proteina alterata chiamata CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) coinvolta nel trasporto transmembrana di sali. L’alterazione di tale proteina provoca un’ anomalia nella secrezione delle ghiandole esocrine con produzione di secrezioni dense e viscose che occludono dotti e canali. La presenza di queste secrezioni anormali determina un danno progressivo degli organi coinvolti: il pancreas si atrofizza, l'intestino si occlude, il fegato trattiene bile, i bronchi si ostruiscono e s'infettano lesionando i polmoni.
 o Mucoviscidosi è una grave malattia congenita, evolutiva e cronica, che colpisce indifferentemente maschi e femmine, trasmessa con meccanismo autosomico recessivo, il cui difetto di base consiste nella produzione di una proteina alterata chiamata CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) coinvolta nel trasporto transmembrana di sali. L’alterazione di tale proteina provoca un’ anomalia nella secrezione delle ghiandole esocrine con produzione di secrezioni dense e viscose che occludono dotti e canali. La presenza di queste secrezioni anormali determina un danno progressivo degli organi coinvolti: il pancreas si atrofizza, l intestino si occlude, il fegato trattiene bile, i bronchi si ostruiscono e s infettano lesionando i polmoni.")
2
FIBROSI CISTICA O MUCOVISCIDOSI
Il gene della Fibrosi Cistica è situato sul braccio lungo del cromosoma 7 e codifica la proteina CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) localizzata nella membrana apicale delle cellule degli epiteli con la funzione di regolare gli scambi idroelettrici. Esistono numerose mutazion, oltre 1000, la più frequente mutazione è la ΔF508 (delezione dell’amminoacido fenilalanina in posizione 508). Le mutazioni FC vengono generalmente distinte in cinque classi principali, a seconda delle alterazioni funzionali e strutturali che determinano sulla proteina: assenza di prodotto proteico; modificazione dei processi biochimici post-traduzionali; alterazione dei meccanismi di regolazione; insufficienza nella funzionalità ionica; riduzione quantitativa dell’mRNA o della proteina.
 localizzata nella membrana apicale delle cellule degli epiteli con la funzione di regolare gli scambi idroelettrici. Esistono numerose mutazion, oltre 1000, la più frequente mutazione è la ΔF508 (delezione dell’amminoacido fenilalanina in posizione 508). Le mutazioni FC vengono generalmente distinte in cinque classi principali, a seconda delle alterazioni funzionali e strutturali che determinano sulla proteina: assenza di prodotto proteico; modificazione dei processi biochimici post-traduzionali; alterazione dei meccanismi di regolazione; insufficienza nella funzionalità ionica; riduzione quantitativa dell’mRNA o della proteina.")
3
FIBROSI CISTICA O MUCOVISCIDOSI
Essa è una delle malattie genetiche più diffuse e interessa circa un nuovo nato su 2500. Una persona su 25 è portatrice sana del gene mutato. Il difetto è localizzato sul cromosoma 7. La malattia si manifesta solo negli omozigoti e due genitori portatori eterozigoti avranno, a livello statistico, il 25% di probabilità di generare un figlio malato (fig. 1). Se in una coppia uno solo dei due genitori (o il padre o la madre) è portatore, ad ogni gravidanza vi è il 50% di probabilità che il figlio sia un portatore e il 50% di probabilità che il figlio non sia portatore (fig. 2). Fig. 1 Fig. 2
. Se in una coppia uno solo dei due genitori (o il padre o la madre) è portatore, ad ogni gravidanza vi è il 50% di probabilità che il figlio sia un portatore e il 50% di probabilità che il figlio non sia portatore (fig. 2). Fig. 1. Fig. 2.")
4
FIBROSI CISTICA O MUCOVISCIDOSI
Sintomatologia La maggior parte dei pazienti affetti da fibrosi cistica presenta i primi sintomi della malattia durante l'infanzia. Circa il 17% dei neonati affetti manifesta, come sintomo della malattia, una ostruzione intestinale, detta ileo da meconio entro le prime 24 ore dalla nascita. Nei casi più comuni la sintomatologia si manifesta con una tosse persistente, broncopolmoniti recidivanti e ritardo nell'accrescimento. Nella maggior parte dei casi le infezioni respiratorie sono dovute allo Staphilococcus aureus, all'Haemophilus influenzae B, allo Pseudomonas Aeruginosa e alla Burkholderi cepacia spesso resistenti ai comuni trattamenti antibiotici.

5
FIBROSI CISTICA O MUCOVISCIDOSI
Sintomatologia Nell‘80-90% dei casi un'insufficienza pancreatica con conseguente malassorbimento proteico e lipidico ed emissione di feci abbondanti ed untuose. Nel fegato il sistema biliare e' interessato nel 15-20% dei casi con ittero da stasi in epoca neonatale e cirrosi epatica focale e multifocale evolvente in insufficienza epatica nell’adulto. Inoltre, a livello del naso e dei seni paranasali, il ristagno di secrezioni e la colonizzazione batterica portano allo sviluppo di una sinusite cronica. Il 95% dei maschi presenta azospermia da ostruzione dei dotti, mentre il 25% delle femmine è infertile a causa dell'alterazione del muco cervicale (muco prodotto e presente a livello della cervice uterina). Tuttavia il 90% delle gravidanze da parte di donne affette è portata a buon fine e di solito le donne affette da fibrosi cistica sono in grado di allattare i propri bambini.
. Tuttavia il 90% delle gravidanze da parte di donne affette è portata a buon fine e di solito le donne affette da fibrosi cistica sono in grado di allattare i propri bambini.")
6
FIBROSI CISTICA O MUCOVISCIDOSI
Un accenno particolare deve essere riservato alla “Sindrome da perdita di sale”, che è un quadro clinico acuto che può presentarsi quando la sudorazione è molto intensa, nei primi anni di vita, durante la stagione estiva, in caso di febbre o in occasione di un’ iperattività fisica e che può condurre ad uno squilibrio metabolico con ipocloremia ed alcalosi.

7
FIBROSI CISTICA O MUCOVISCIDOSI
DIAGNOSI CLINICA E MOLECOLARE La diagnosi di FC allo stato attuale delle conoscenze può essere sospettata e formulata in presenza di: uno o più sintomi evocativi o compatibili con la malattia; 2. diagnosi di FC accertata in un fratello o in parente consanguineo; 3. screening neonatale per la FC positivo; presenza di due mutazioni associate a FC; test del sudore positivo.

8
FIBROSI CISTICA O MUCOVISCIDOSI
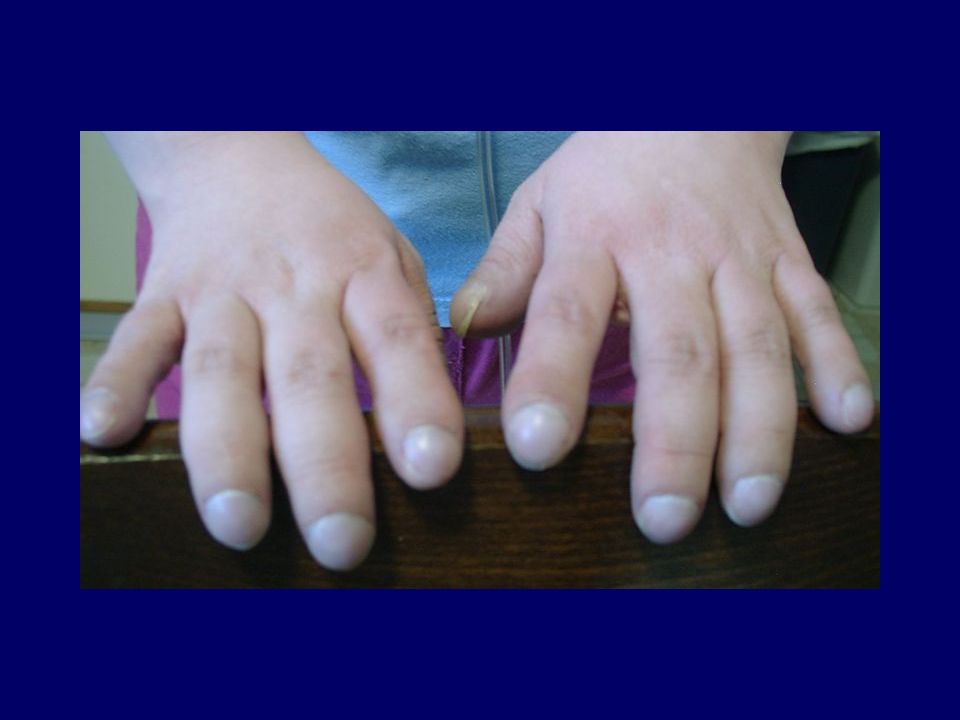
Sintomi evocativi di F.C. La Fibrosi Cistica deve essere sospettata nei lattanti quando presentano: ittero colostatico neonatale protratto; diarrea persistente con feci abbondanti e fetide; scarso accrescimento a dispetto di un vorace appetito; sindrome da perdita di sali (shock ipovolemico con ipocloremia ed alcalosi metabolica); tosse pertussoide. In seguito a qualunque età, la Fibrosi Cistica deve essere sospettata in pazienti che presentino: dolori addominali intensi, ricorrenti (occlusione o subocclusione intestinale) senza causa apparente; tosse catarrale ricorrente o cronica; torace deformato “a botte” e/o ippocratismo digitale; precedenti diagnosi di tubercolosi e intradermoreazione negativa; emottisi sia massiva che modesta, specie se ricorrente; pneumotorace spontaneo.
; tosse pertussoide. In seguito a qualunque età, la Fibrosi Cistica deve essere sospettata in pazienti che presentino: dolori addominali intensi, ricorrenti (occlusione o subocclusione intestinale) senza causa apparente; tosse catarrale ricorrente o cronica; torace deformato a botte e/o ippocratismo digitale; precedenti diagnosi di tubercolosi e intradermoreazione negativa; emottisi sia massiva che modesta, specie se ricorrente; pneumotorace spontaneo.")
10
FIBROSI CISTICA O MUCOVISCIDOSI
Diagnosi La diagnosi può essere "sospettata" sin dalla nascita in caso di ileo da meconio. E’ possibile effettuare il dosaggio della tripsina nel sangue prelevato al quarto giorno di vita. Test del sudore con il quale vendono dosati i livelli di Cloro e Sodio che sono tipicamente aumentati. La certezza viene dall'identificazione delle mutazioni geniche. Oggi l'analisi genetica consente di riconoscere l'85% dei portatori. Nelle famiglie a rischio, cioè nelle famiglie in cui i genitori sono portatori sani è possibile la diagnosi prenatale nel primo trimestre di gravidanza. Normale Fibrosi cistica Osmolarità ± Na+ (nM) Cl- (nM) K+ (nM)
 Cl- (nM) K+ (nM)")
11
FIBROSI CISTICA O MUCOVISCIDOSI
TERAPIA E' rivolta a contrastare l'evoluzione della malattia e si basa su: fisioterapia respiratoria dieta equilibrata molto ricca di Sali somministrazione di enzimi pancreatici ad ogni pasto antibioticoterapia nei confronti dei germi isolati dalle secrezioni bronchiali terapia medica delle complicanze nei casi più gravi, trapianto di polmoni e/o fegato Questi interventi hanno consentito di ottenere aspettative di vita superiori a quelle del passato quando di fibrosi cistica si moriva nei primi anni di vita. La vita media si avvicina ai 40 anni, ma le cure agiscono sui sintomi e non sul difetto di base della malattia. Sono allo studio nuovi farmaci, attualmente in fase di sperimentazione clinica.



















>")


