Scaricare la presentazione
1
LEUCEMIE Leucemia: sotto questo nome rientrano tutti i tumori dei globuli bianchi (leucociti), che sono una delle popolazioni di cellule circolanti nel sangue. Data la loro origine questi tumori non sono solidi, cioè non sono caratterizzati dalla presenza di una massa tumorale, ma dalla proliferazione incontrollata di queste cellule che invadono tessuti e organi. Nel sangue umano esistono tre diversi gruppi di globuli bianchi (linfociti, granulociti e monociti), quindi sono possibili tre diversi tipi di leucemia: leucemia linfoide (coinvolge la produzione di linfociti) leucemia mieloide (coinvolge la produzione di granulociti) leucemia monocitica (che coinvolge la produzione di monociti) La leucemia si divide inoltre sulla base del decorso clinico in leucemia acuta (con una evoluzione rapida) e leucemia cronica.
, che sono una delle popolazioni di cellule circolanti nel sangue. Data la loro origine questi tumori non sono solidi, cioè non sono caratterizzati dalla presenza di una massa tumorale, ma dalla proliferazione incontrollata di queste cellule che invadono tessuti e organi. Nel sangue umano esistono tre diversi gruppi di globuli bianchi (linfociti, granulociti e monociti), quindi sono possibili tre diversi tipi di leucemia: leucemia linfoide (coinvolge la produzione di linfociti) leucemia mieloide (coinvolge la produzione di granulociti) leucemia monocitica (che coinvolge la produzione di monociti) La leucemia si divide inoltre sulla base del decorso clinico in leucemia acuta (con una evoluzione rapida) e leucemia cronica.")
2
LEUCEMIA ACUTA LINFOBLASTICA
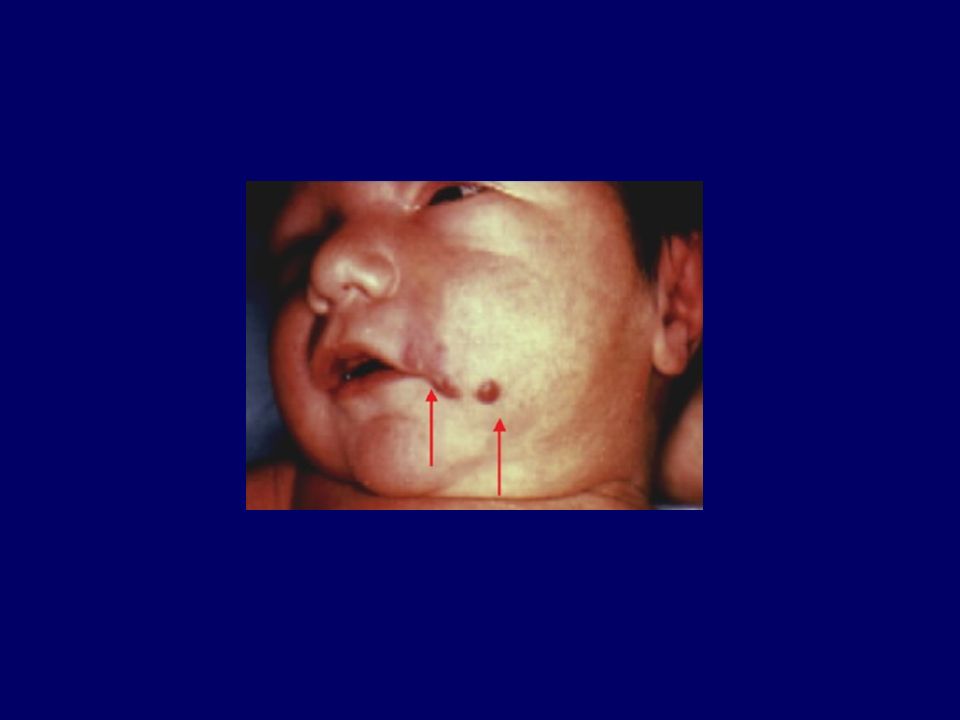
Malattia linfoproliferativa originata da un primitivo disordine dell’emopoiesi che si manifesta con una produzione rapida ed incontrollata di precursori della linea linfoide. Epidemiologia: 28-36 nuovi casi/anno/ di soggetti di età < 15 anni ( nuovi casi/anno in Italia) picco d’incidenza: 3-5 anni 75-80% delle leucemie dell’età pediatrica eziologia sconosciuta, anche se vengono chiamati in causa fattori predisponenti di natura genetica, virale, immunologica ed ambientale. Principali dati clinici: Anoressia, astenia, pallore cutaneo, febbre, petecchie, dolori osteoarticolari, epatosplenomegalia, linfoadenomegalia.
 picco d’incidenza: 3-5 anni % delle leucemie dell’età pediatrica. eziologia sconosciuta, anche se vengono chiamati in causa fattori predisponenti di natura genetica, virale, immunologica ed ambientale. Principali dati clinici: Anoressia, astenia, pallore cutaneo, febbre, petecchie, dolori osteoarticolari, epatosplenomegalia, linfoadenomegalia.")
3
LEUCEMIA ACUTA LINFOBLASTICA
Classificazione morfologica secondo il gruppo FAB L1 Linfoblasti con scarso citoplasma e nucleoli poco rappresentati (85% dei pazienti con LLA). L2 Linfoblasti generalmente più grandi, anche se di grandezza variabile con citoplasma e nucleoli più grandi (14% dei pazienti con LLA). L3 Linfoblasti grandi, omogenei, notevolmente basofili, abbondanti, con citoplasma vacuolato e nucleoli vescicolari. Caratteristiche identiche a quelle delle cellule di Burkitt (1% dei pazienti con LLA).
. L2 Linfoblasti generalmente più grandi, anche se di grandezza variabile con citoplasma e nucleoli più grandi (14% dei pazienti con LLA). L3 Linfoblasti grandi, omogenei, notevolmente basofili, abbondanti, con citoplasma vacuolato e nucleoli vescicolari. Caratteristiche identiche a quelle delle cellule di Burkitt (1% dei pazienti con LLA).")
4
LEUCEMIA ACUTA LINFOBLASTICA
DIAGNOSI Esame sangue periferico Aspirato midollare 25-90% di cellule blastiche anemia normocromica normocitica piastrinopenia globuli bianchi , raramente linfociti atipici (linfoblasti) caratterizzazione citochimica: PAS +, perossidasi -, sudan -, Esterasi -, Fosfatasi Acida + caratterizzazione citomorfologica (sec FAB): L1, L2, L3 fenotipo immunologico: B (pre-preB, preB, B matura, Common Acute Lymphoblastic Leukemia Antigen - CALLA) T (pre-T. T intermedia, T matura) Citogenetica (traslocazione): t(9;22), t(4;11), t(12;21), t(1;19) studio molecolare
 caratterizzazione citochimica: PAS +, perossidasi -, sudan -, Esterasi -, Fosfatasi Acida + caratterizzazione citomorfologica (sec FAB): L1, L2, L3. fenotipo immunologico: B (pre-preB, preB, B matura, Common Acute Lymphoblastic Leukemia Antigen - CALLA) T (pre-T. T intermedia, T matura) Citogenetica (traslocazione): t(9;22), t(4;11), t(12;21), t(1;19) studio molecolare.")
5
LLA - TRATTAMENTO Il cardine della cura è la chemioterapia, sia nei bambini sia negli adulti. Questa è somministrata per via endovenosa, eventualmente attraverso un catetere venoso centrale. Alcuni farmaci possono essere somministrati per os o per via intramuscolare. La chemioterapia viene somministrata in tre-quattro fasi: Induzione della remissione Terapia post remissione (a volte suddivisa in fase di consolidamento e fase di intensificazione della remissione) Terapia di mantenimento
 Terapia di mantenimento.")
6
LLA - TRATTAMENTO Con la TERAPIA DI INDUZIONE si cerca di ottenere la remissione completa, ottenibile nel 90-95% dei casi delle leucemia linfoblastica acuta dei bambini, di meno negli adulti. Per REMISSIONE COMPLETA si intende il ripristino delle condizioni normali nel midollo e nel sangue periferico: meno del 5% di blasti nel sangue periferico ed assenza di anemia, piastrinopenia, leucocitosi e/o leucopenia. Ottenere la remissione completa rappresenta indubbiamente il primo, fondamentale, importante passo verso la guarigione definitiva. Tuttavia non abbiamo oggi a disposizione un mezzo sicuro che ci consenta di stabilire se dopo questa prima fase di terapia siano state distrutte tutte le cellule leucemiche. È per questo motivo che si esegue sempre la terapia post remissione con la speranza che, continuando la terapia, si riesca ad eliminare anche l’ultima cellula eventualmente rimasta. La TERAPIA DI MANTENIMENTO consiste nella continuazione della terapia per un periodo di circa due-tre anni dopo l’ottenimento della remissione completa e ha lo scopo di mantenere la remissione eliminando eventuali cellule rimaste. Il 70-75% dei bambini con leucemia linfoblastica acuta che ottengono la remissione completa sono lungo sopravviventi e possono essere considerati guariti definitivamente (malattia residua minima!!). Le percentuali sono inferiori negli adulti.
. Le percentuali sono inferiori negli adulti.")
7
COMPLICANZE DELLA TERAPIA
LLA - TRATTAMENTO I farmaci più utilizzati per indurre la remissione sono: la vincristina, i cortisonici, la daunorubicina e la L-asparaginasi. Nella fase postremissione è probabile che vengano usati altri farmaci come l’etoposide, la citarabina. Nella fase di mantenimento sono utilizzati in genere il methotrexate e la 6-mercaptopurina, che possono essere somministrati per os. COMPLICANZE DELLA TERAPIA SINDROME DELLA LISI TUMORALE: è legata ad uno squilibrio metabolico dovuto alla rapida lisi delle cellule leucemiche con rilascio di fosfato, acido urico e di potassio ad una velocità che eccede la clearance renale → iperkalemia, iperfosfatemia, ipocalcemia (ipoparatiroidismo!!) ed iperuricemia. MIELOSOPPRESSIONE: espone il bambino al rischio di infezioni soprattutto da parte di Stafilococchi, Gram negativi, virus e funghi. E’ consigliabile terapia antibiotica a largo spettro. EFFETTI COLLATERALI A LUNGO TERMINE DI NATURA NEUROTOSSICA LEGATI ALLA RADIOTERAPIA:diminuzione QI, atassia, convulsioni e disfunzione intellettiva.
 ed iperuricemia. MIELOSOPPRESSIONE: espone il bambino al rischio di infezioni soprattutto da parte di Stafilococchi, Gram negativi, virus e funghi. E’ consigliabile terapia antibiotica a largo spettro. EFFETTI COLLATERALI A LUNGO TERMINE DI NATURA NEUROTOSSICA LEGATI ALLA RADIOTERAPIA:diminuzione QI, atassia, convulsioni e disfunzione intellettiva.")
8
LEUCEMIA ACUTA MIELOBLASTICA
Gruppo di sindromi mieloproliferative acute caratterizzate da un deficit delle capacità differenziative e maturative della cellula staminale mieloide. Epidemiologia: 5 -10 nuovi casi/anno/ di soggetti di età < 15 anni 15% delle leucemie acute dell’età pediatrica Eziologia sconosciuta. Forme secondarie a trattamento chemio e/o radioterapico per altre neoplasie Principali dati clinici: Anoressia, astenia, pallore cutaneo, febbre, perdita di peso, epatosplenomegalia, emorragie cutanee e mucose, linfoadenomegalia, ipertrofia gengivale (folati!!).
.")
10
LEUCEMIA ACUTA MIELOBLASTICA 30-90% di cellule blastiche
DIAGNOSI Aspirato midollare 30-90% di cellule blastiche Esame sangue periferico anemia normocromica normocitica piastrinopenia globuli bianchi , o normali presenza di blasti e granulociti senza precursori intermedi (hiatus leucemico) caratterizzazione citochimica: perossidasi +, PAS+, alfa-naftil-acetato-esterasi +, N-ASD-cloroacetato-esterasi +, sudan nero + caratterizzazione citomorfologica (sec. FAB): M1, M2, M3, M4, M5, M6, M7, M0 caratterizzazione immunologica caratterizzazione molecolare caratterizzazione citogenetica: M1 t (9;22) M4 11q23 M4eo inv 16 – t (16;16) M2 t(8;21) M5 11q23 M3 t (15;17) t (11;17) M7 t(1;22)
 caratterizzazione citochimica: perossidasi +, PAS+, alfa-naftil-acetato-esterasi +, N-ASD-cloroacetato-esterasi +, sudan nero + caratterizzazione citomorfologica (sec. FAB): M1, M2, M3, M4, M5, M6, M7, M0. caratterizzazione immunologica. caratterizzazione molecolare. caratterizzazione citogenetica: M1 t (9;22) M4 11q23. M4eo inv 16 – t (16;16) M2 t(8;21) M5 11q23. M3 t (15;17) t (11;17) M7 t(1;22)")
11
LINFOMI NON HODGKIN (LNH)
Gruppo eterogeneo di neoplasie che originano dalla degenerazione maligna di cellule appartenenti a linee diverse del sistema immunitario. Epidemiologia: 3-4 nuovi casi/anno/ di soggetti di età < 15 anni picco d’incidenza: 7 – 11 anni M/F = 3:1 eziologia sconosciuta

12
LINFOMI NON HODGKIN (LNH)
PRINCIPALI DATI CLINICI Interessamento mediastinico Tosse, febbricola, dispnea ingravescente, versamento pleurico, sindrome della vena cava superiore (compressione!!). Interessamento addominale Dolori addominali, vomito, massa addominale, quadro di addome acuto. Interessamento linfonodale Linfoadenopatia laterocervicale, più raramente ascellare o inguinale.
. Interessamento addominale. Dolori addominali, vomito, massa addominale, quadro di addome acuto. Interessamento linfonodale. Linfoadenopatia laterocervicale, più raramente ascellare o inguinale.")
13
LINFOMI NON HODGKIN (LNH)
istologica aspirato midollare esame citologico (versamento pleurico o ascitico) Diagnosi: Anatomia patologica: Stadiazione: Linfomi maligni ad alto grado di malignità secondo la class. REAL: linfoma maligno (a grandi cellule, immunoblastico, plasmocitoide, a cellule chiare, polimorfo, con componente a cellule epitelioidi); linfoma maligno linfoblastico (a cellule convolute, a cellule non convolute); linfoma maligno a piccole cellule non clivate (linfoma di Burkitt, con aree follicolari) Fenotipo immunologico: T, B, nonT nonB Clinico-strumentale; chirurgica solo in caso di LNH addominale; classificazione in stadi secondo Murphy (I-IV)
 Diagnosi: Anatomia patologica: Stadiazione: Linfomi maligni ad alto grado di malignità secondo la class. REAL: linfoma maligno (a grandi cellule, immunoblastico, plasmocitoide, a cellule chiare, polimorfo, con componente a cellule epitelioidi); linfoma maligno linfoblastico (a cellule convolute, a cellule non convolute); linfoma maligno a piccole cellule non clivate (linfoma di Burkitt, con aree follicolari) Fenotipo immunologico: T, B, nonT nonB. Clinico-strumentale; chirurgica solo in caso di LNH addominale; classificazione in stadi secondo Murphy (I-IV)")
14
MALATTIA DI HODGKIN Processo neoplastico di verosimile origine linfoide o monocito-macrofagica contrassegnato da un tipico marker citologico: la cellula di Reed-Sternberg. Epidemiologia: 5 - 6 nuovi casi/anno/ di soggetti di età < 15 anni picco d’incidenza: seconda decade M/F = 2.3:1 eziologia sconosciuta Principali dati clinici: Linfoadenomegalia laterocervicale e/o sopraclaveare. I linfonodi coinvolti sono gommosi, non dolenti, isolati o raggruppati o circondati da linfonodi normalmente reattivi. Segni sistemici: febbre, sudorazione notturna, calo ponderale.

15
STAGING DEI LINFOMI DI HODGKIN
STADIO I: Coinvolgimento di una singola regione linfonodale o di una singola sede extralinfatica. STADIO II: Coinvolgimento di 2 o più regioni linfonodali nello stesso lato del diaframma oppure coinvolgimento di una sede extralinfatica ed una o più regioni linfonodali sullo stesso lato del diaframma. STADIO III: Coinvolgimento di regioni linfonodali in entrambi i lati del diaframma con possibile coinvolgimento localizzato di un organo o di una sede extralinfatica e/o coinvolgimento della milza. STADIO IV: Coinvolgimento disseminato di uno o più organi o tessuti extralinfatici con o senza ingrossamento linfonodale..

16
TUMORI del S.N.C. Epidemiologia: Principali dati clinici:
Gruppo eterogeneo di neoplasie maligne per sede e tipo istologico. Epidemiologia: nuovi casi/anno/ di soggetti di età < 15 anni 21% di tutte le neoplasie dell’età pediatrica Picco d’incidenza: 5 – 10 anni eziologia sconosciuta Principali dati clinici: Segni di ipertensione endocranica Cefalea, vomito a getto, papilledema, strabismo, diplopia; nei pz < 2 anni: accresciuta irritabilità, circonferenza cranica, allargamento e tensione della fontanella, stasi delle vene del cuoio capelluto, occhi a “sole calante”.

17
TUMORI del S.N.C. Principali segni clinici:
T. cervelletto e IV ventricolo Astenia, asinergia, nistagmo, ipotonia muscolare, torcicollo o rigidità nucale T. del tronco cerebrale Paralisi progressive multiple e bilaterali a carico dei nervi cranici, atassia T. emisferi e ventricoli laterali Paresi, episodi comizali T. sellari, parasellari o soprasellari Ritardo di crescita (o pubertà ritardata e obesità), progressiva perdita del visus, diabete insipido
, progressiva perdita del visus, diabete insipido.")
18
TUMORI del S.N.C. Diagnosi: Anatomia patologica:
Istologica: prelievo bioptico mediante stereotassi della sede interessata, quando possibile Strumentale: TAC, RMN, angiografia Anatomia patologica: T. sopratentoriali: gliomi, craniofaringiomi, ependimomi, adenomi, pinealomi, gliomi ottici T. sottotentoriali: medulloblastomi, astrocitomi cerebellari, ependimomi, gliomi, peduncoli cerebrali

19
NEUROBLASTOMA Neoplasia maligna ad insorgenza dalle cellule della cresta neurale Epidemiologia: 6.5 – 10.1 nuovi casi/anno/ di soggetti di età < 15 anni picco d’incidenza: 0 – 2 anni M/F = 1.2:1 eziologia sconosciuta

20
NEUROBLASTOMA Principali dati clinici Localizzazione addominale
Vaghi dolori addominali, anoressia, vomito, massa palpabile nei quadranti laterali dell’addome o in regione alto costale, abitualmente oltrepassante la linea mediana. Localizzazione intratoracica Tosse, dispnea, disfagia, segni compressivi a carico del midollo spinale. Segni sistemici Dolore osteo-articolari, astenia, perdita di peso, pallore, febbre ricorrente.

21
NEUROBLASTOMA Diagnosi: istologica
aspirato midollare e/o biopsia ossea VMA (acido vanil mandelico), HVA (acido omovanillico) Neuroblastoma, ganglioneuroblastoma, ganglioneuroma, feocromocitoma Anatomia patologica: Stadiazione: Chirurgica e clinico-strumentale Classificazione in stadi sec. INSS (I-IVs) o in gruppi sec. AIEOP (I-V)
, HVA (acido omovanillico) Neuroblastoma, ganglioneuroblastoma, ganglioneuroma, feocromocitoma. Anatomia patologica: Stadiazione: Chirurgica e clinico-strumentale. Classificazione in stadi sec. INSS (I-IVs) o in gruppi sec. AIEOP (I-V)")
22
TUMORE DI WILMS Neoplasia maligna di tipo embrionario Epidemiologia:
5.5 – 7.1 nuovi casi/anno/ di soggetti di età < 15 anni 5-10% di tutti i tumori solidi dell’età pediatrica picco d’incidenza: 1-5 anni M/F = 0.9:2 alterazioni gene WT1 (banda 11p13) situato sul braccio corto del cromosoma 11 forma ereditaria (15-20%), bilaterale o multifocale, spesso associata ad altre anomalie congenite (aniridia, emi-ipertrofia corporea, anomalie del tratto genito-urinario) forma sporadica
 situato sul braccio corto del cromosoma 11. forma ereditaria (15-20%), bilaterale o multifocale, spesso associata ad altre anomalie congenite (aniridia, emi-ipertrofia corporea, anomalie del tratto genito-urinario) forma sporadica.")
23
TUMORE DI WILMS Principali dati clinici:
Fortuito riscontro di “massa addominale” non oltrepassante la linea mediana, macroematuria, ipertensione, stipsi, anoressia, febbre, vaghi dolori addominali. Diagnosi: istologica Anatomia patologica: anaplastico, sarcomatoso Chirurgica e anatomo.patologica basata sul grado di estensione macro o microscopica del tumore (stadio I-V sec. NWTS) Stadiazione:
 Stadiazione:")
24
SARCOMA DI EWING Epidemiologia:
Tumore maligno primitivo dell’osso, caratterizzato da tessuto d’aspetto istologico uniforme rappresentato da cellule piccole e rotonde con scarso citoplasma. La famiglia dei tumori di Ewing comprende: il tumore di Ewing dell’osso (ETB), il tumore di Ewing extraosseo (EOE), il tumore primitivo neuroectodermico (PNET) ed il tumore di Askin (PNET della parete toracica). Epidemiologia: 1.7 – 2.1 nuovi casi/anno/ di soggetti di età < 20 anni picco d’incidenza: anni maggiore frequenza nel sesso maschile 4% dei tumori maligni dell’infanzia ed il 10-15% di tutti i tumori ossei dell’età pediatrica
, il tumore di Ewing extraosseo (EOE), il tumore primitivo neuroectodermico (PNET) ed il tumore di Askin (PNET della parete toracica). Epidemiologia: 1.7 – 2.1 nuovi casi/anno/ di soggetti di età < 20 anni. picco d’incidenza: anni. maggiore frequenza nel sesso maschile. 4% dei tumori maligni dell’infanzia ed il 10-15% di tutti i tumori ossei dell’età pediatrica.")
25
SARCOMA DI EWING Principali dati clinici:
Febbre, dolore, tumefazione a carico del segmento osseo colpito, e dei tessuti molli circostanti; esordiscono tipicamente a carico della pelvi, degli arti, delle regioni paraspinali della parete toracica e metastatizzano alle ossa, al midollo osseo e ai polmoni. Diagnosi: radiologica istologica Anatomia patologica: quadro privo di caratteristiche peculiari

26
RABDOMIOSARCOMA Neoplasia maligna ad insorgenza dalle cellule del muscolo scheletrico che rappresenta circa il 50% dei sarcomi dei tessuti molli del bambino. Epidemiologia: 3.7 – 5 nuovi casi/anno/ di soggetti di età < 20 anni picco d’incidenza: 2-5 anni M/F = 1.4:1 eziologia sconosciuta Rare forme ereditarie associate a mutazioni del oncogene soppressore p53 (Sindrome di Li-Fraumeni)
")
27
RABDOMIOSARCOMA Principali dati clinici: Testa e collo
Disfonia, disfagia, tumefazione a carico dei tessuti molli, sinusite, epistassi, esoftalmo. Tronco ed estremità Massa palpabile, sintomatologia compressiva a carico del midollo spinale. Tratto genito-urinario Disturbi minzionali, ematuria, sanguinamento vaginale. Diagnosi: istologica

28
LOCALIZZAZIONE CARATTERISTICA
RABDOMIOSARCOMA Anatomia patologica: ISTOTIPO LOCALIZZAZIONE CARATTERISTICA embrionale (60-70%) distretto testa-collo tratto genito-urinario alveolare (20%) estremità – tronco – regione perineale - perirettale botrioide (10%) sotto la superficie della mucosa di vescica, vagina e narici pleomorfo (raro) nessuna IRSG suddivide i pazienti in basso, intermedio ed alto rischio combinando il gruppo clinico di appartenenza (I-IV) e stadiazione. Stadiazione:
 distretto testa-collo. tratto genito-urinario. alveolare (20%) estremità – tronco – regione perineale - perirettale. botrioide (10%) sotto la superficie della mucosa di vescica, vagina e narici. pleomorfo (raro) nessuna. IRSG suddivide i pazienti in basso, intermedio ed alto rischio combinando il gruppo clinico di appartenenza (I-IV) e stadiazione. Stadiazione:")
29
OSTEOSARCOMA Neoplasia primitiva dell’osso, caratterizzato dalla formazione diretta di osso o di tessuto osteoide da parte di cellule tumorali. Epidemiologia: 4– 6 nuovi casi/anno/ di soggetti di età < 15 anni 5% dei tumori solidi dell’età pediatrica massima incidenza 15 anni (M), 14 anni (F) eziologia sconosciuta; predisposizione genetica a sviluppare la malattia soprattutto nei pazienti affetti da retinoblastoma ereditario (gene RB1 individuato a livello del locus q14 del cromosoma 13 la cui assenza in entrambi gli alleli predispone ad entrambe le patologie).
, 14 anni (F) eziologia sconosciuta; predisposizione genetica a sviluppare la malattia soprattutto nei pazienti affetti da retinoblastoma ereditario (gene RB1 individuato a livello del locus q14 del cromosoma 13 la cui assenza in entrambi gli alleli predispone ad entrambe le patologie).")
30
OSTEOSARCOMA Principali dati clinici:
Dolore e tumefazione a livello del segmento osseo colpito: femore, tibia, omero, mandibola, mascella, fibula. Polmone: sede elettiva di metastasi Diagnosi: radiologica (TAC, RMN, Scintigrafia con TC99m MDP) istologica: osteoblastico, condroblastico, fibroblastico, parostale, priostiale Stadiazione: Localizzato: estensione limitata all’osso di origine + metastasi nello stesso osso (prognosi peggiore) Metastatico: presenza di metastasi al polmone o ad altri segmenti ossei, o sedi extra-ossee
 istologica: osteoblastico, condroblastico, fibroblastico, parostale, priostiale. Stadiazione: Localizzato: estensione limitata all’osso di origine + metastasi nello stesso osso (prognosi peggiore) Metastatico: presenza di metastasi al polmone o ad altri segmenti ossei, o sedi extra-ossee.")














>")





