Scaricare la presentazione
1
Accrescimento e bassa statura
M. Caruso Dipartimento di Pediatria Università di Catania Azienda Ospedaliero Universitaria Policlinico-Vittorio Emanuele

2
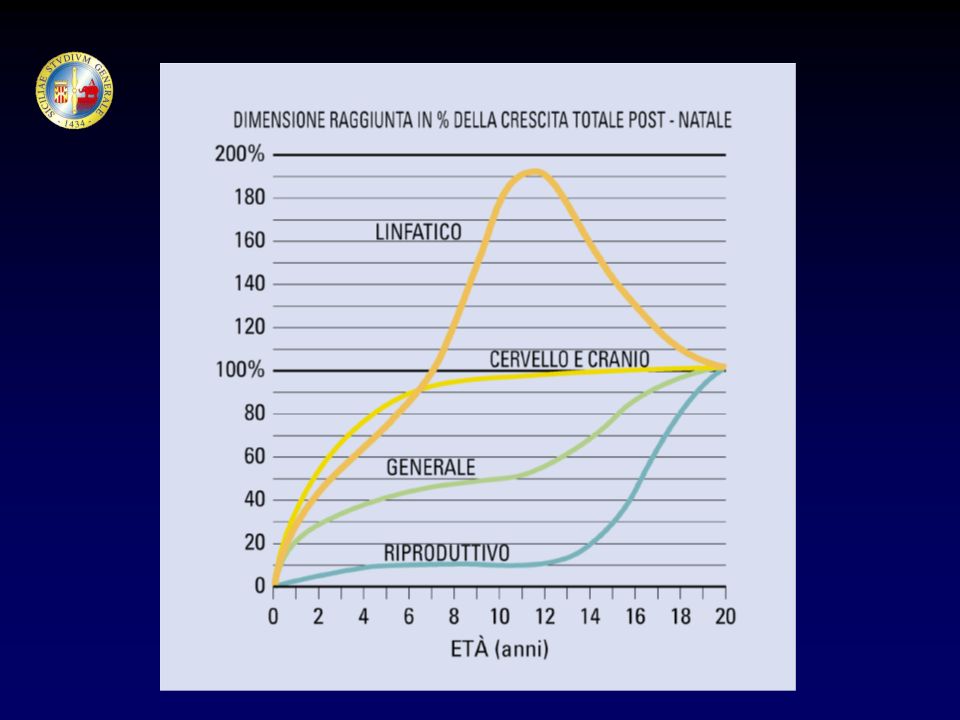
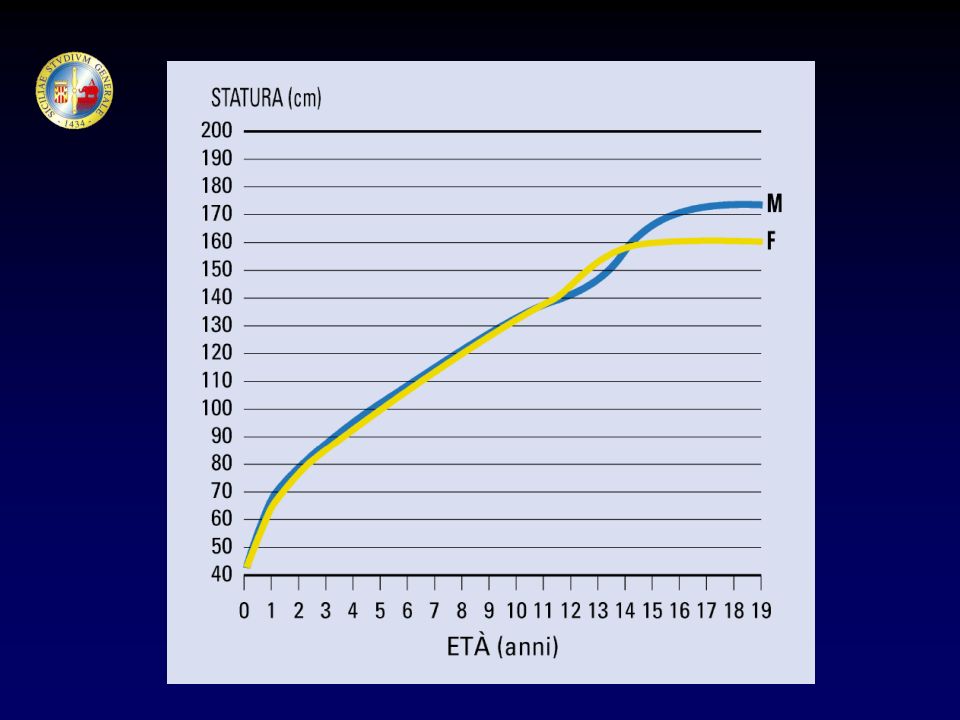
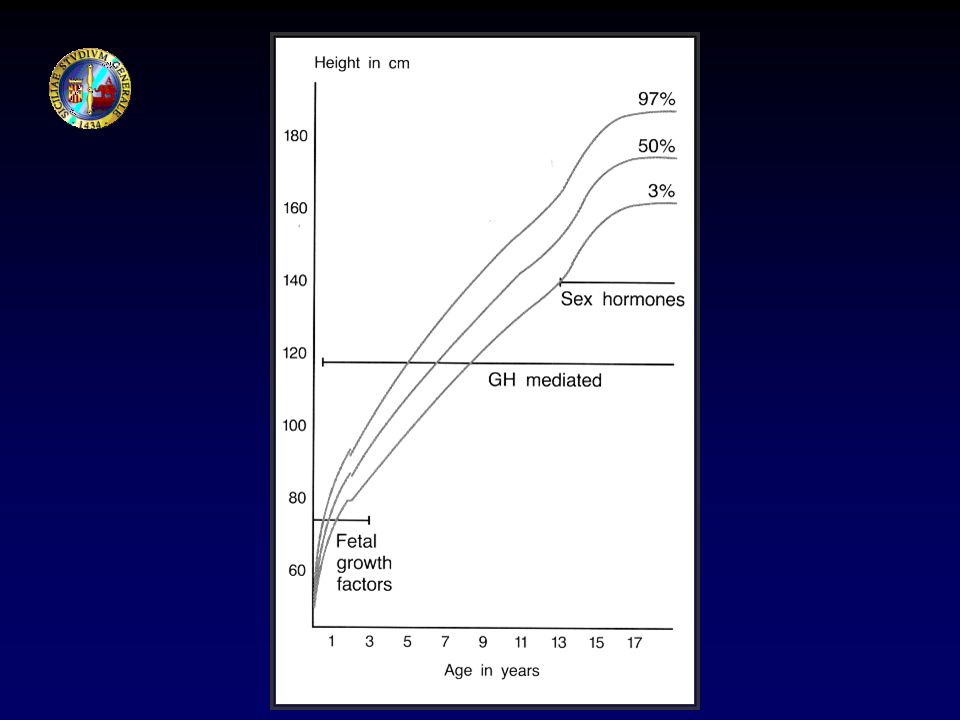
CRESCITA SVILUPPO MATURAZIONE EVOLUZIONE Neonato (1-30 giorni)
I infanzia (1-2 anni) II infanzia (2-6 anni) III infanzia (6 anni - pubertà) Pubertà Adolescenza Età adulta CRESCITA SVILUPPO MATURAZIONE EVOLUZIONE
 II infanzia (2-6 anni) III infanzia (6 anni - pubertà) Pubertà. Adolescenza. Età adulta. CRESCITA. SVILUPPO. MATURAZIONE. EVOLUZIONE.")
8
FATTORI CHE INFLUENZANO LA CRESCITA
F. genetici F. nutrizionali F. ormonali F. ambientali

9
GENETICA DELLA CRESCITA
La correlazione tra statura adulta del soggetto e quella dei consanguinei aumenta con il grado di parentela: 100% gemelli omozigoti → 25% parenti di 2° grado Ereditarietà di tipo poligenico Effetto additivo di geni localizzati su autosomi ed eterocromosomi

10
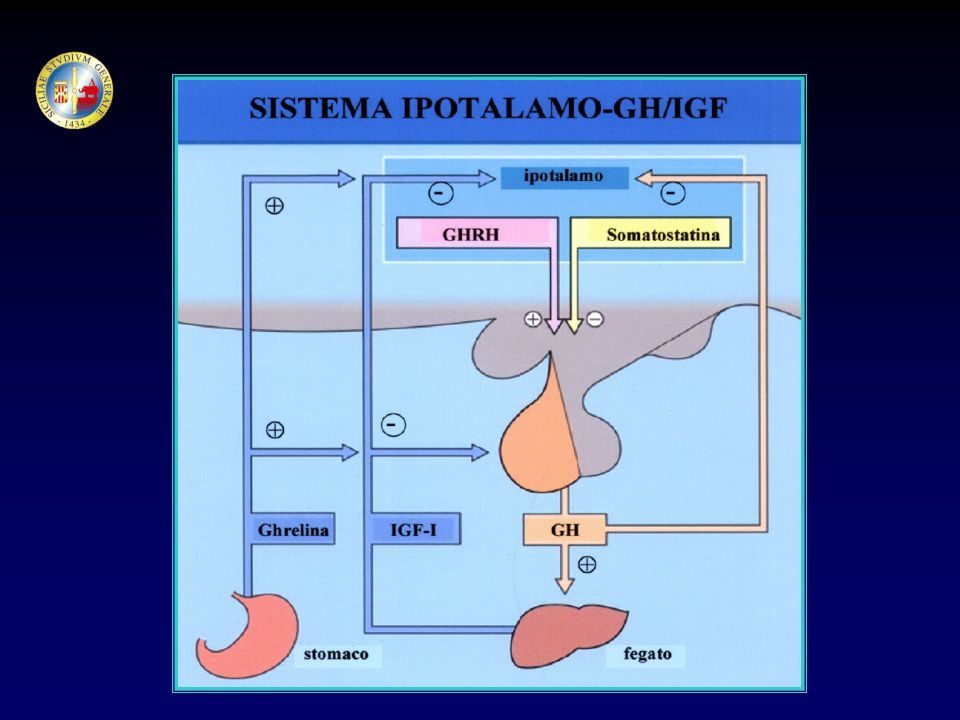
FATTORI ORMONALI GH → IGF-1, IGF-BP Ormoni tiroidei Cortisolo Insulina
Ormoni sessuali

12
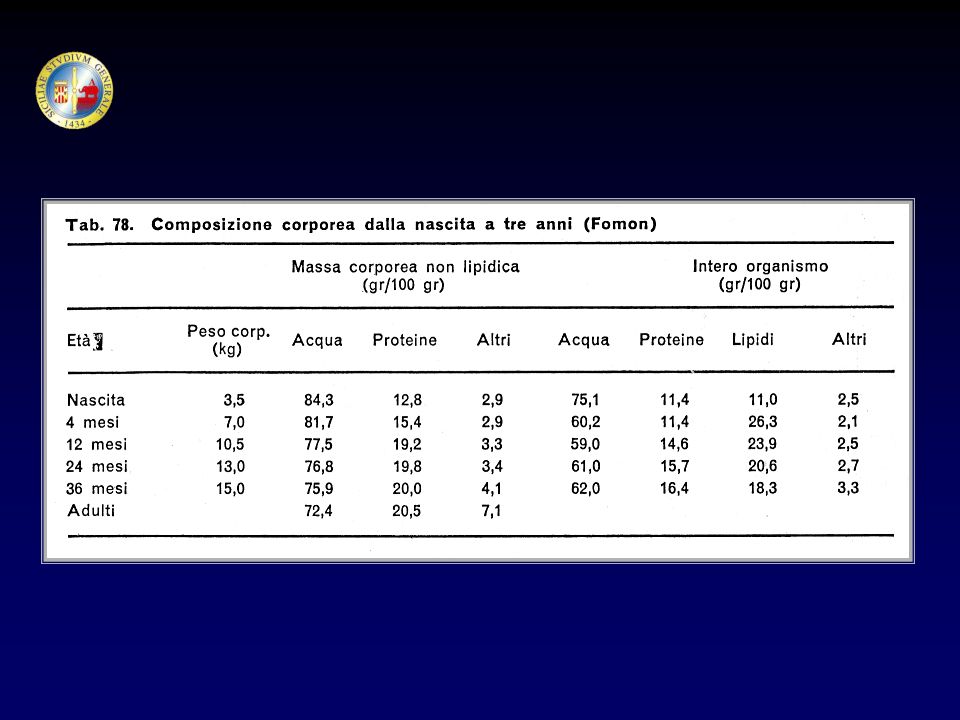
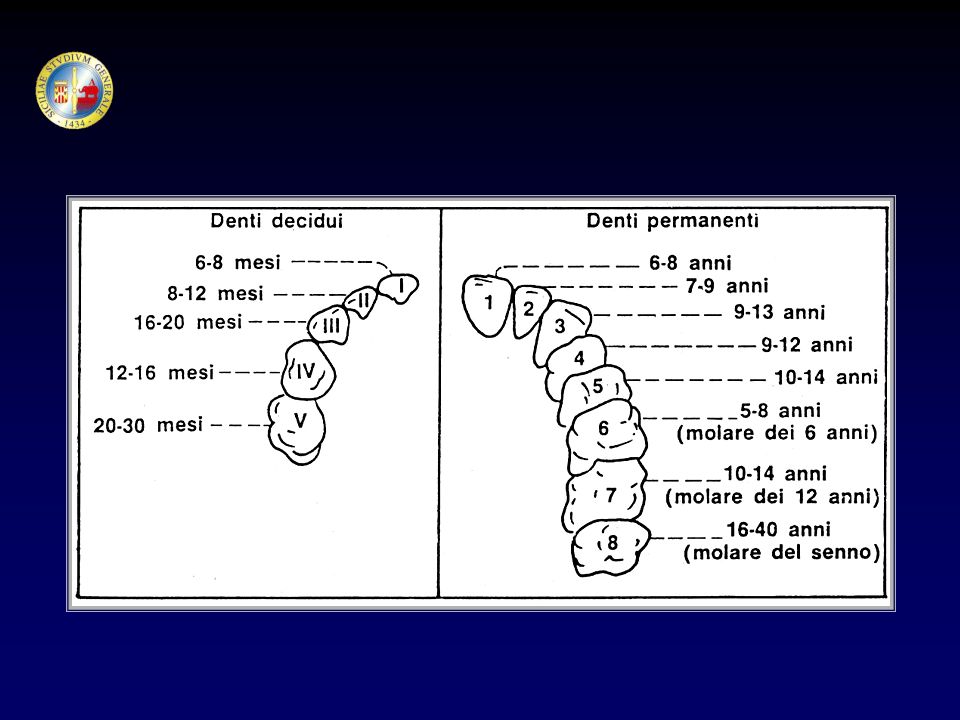
Le ossa nel feto sono inizialmente costituite da tessuto cartilagineo.
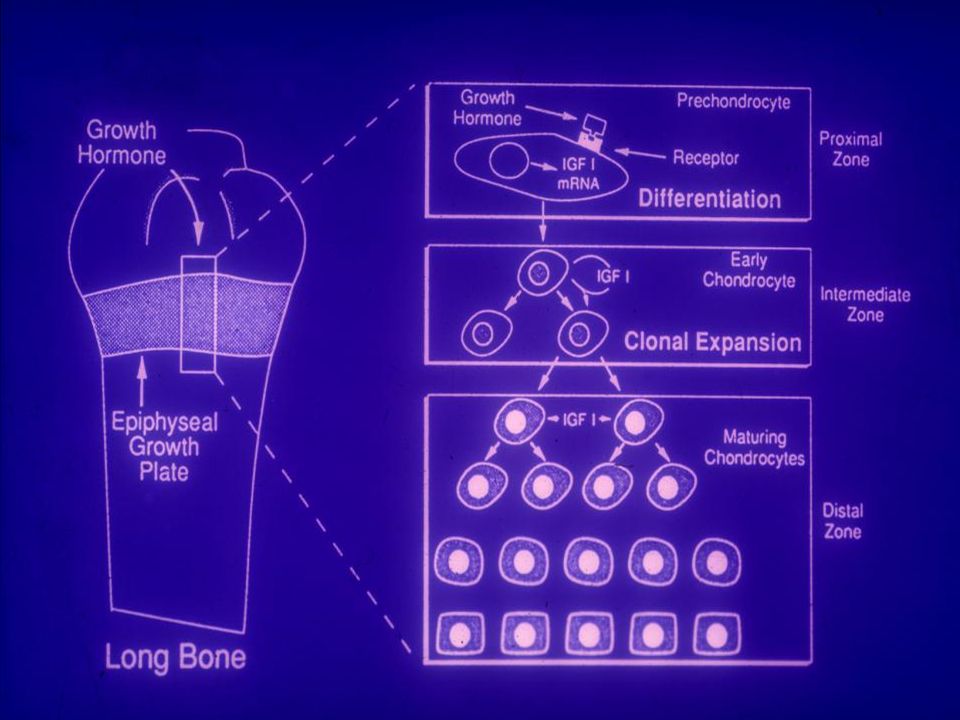
La CRESCITA OSSEA Le ossa nel feto sono inizialmente costituite da tessuto cartilagineo. Il tessuto cartilagineo si converte in tessuto osseo per apposizione di Sali di calcio. La successiva crescita in lunghezza è dovuta alla formazione di cartilagine che viene gradualmente sostituita da osso. L’allungamento delle ossa degli arti (ossa lunghe) avviene per un processo di formazione ossea di particolari siti definiti “di ossificazione”, che si trovano alle 2 estremità dell’osso (epifisi); prima della pubertà le epifisi sono separate dalla parte principale dell’osso (diafisi) da uno strato di cartilagine e di cellule cartilaginee (condrociti).
 avviene per un processo di formazione ossea di particolari siti definiti di ossificazione , che si trovano alle 2 estremità dell’osso (epifisi); prima della pubertà le epifisi sono separate dalla parte principale dell’osso (diafisi) da uno strato di cartilagine e di cellule cartilaginee (condrociti).")
13
LA CRESCITA OSSEA Condrociti Epifisi Cartilagine Diafisi Metafisi
Siti di ossificazione Epifisi Condrociti

14
LA CRESCITA OSSEA Lo strato di condrociti è chiamato cartilagine di accrescimento I condrociti si dividono in senso verticale e le cellule cartilaginee più vecchie vengono spinte verso il basso. Quando queste cellule muoiono, lo spazio da loro occupato si vuota e viene invaso dalle cellule che formano l’osso: osteoblasti ed osteociti che incorporando Sali di calcio trasformano questa porzione in osso. L’allungamento dell’osso cessa quando la cartilagine di accrescimento è sostituito dall’osso e l’epifisi si fonde con la diafisi. Età: 16 anni per le ragazze anni per i ragazzi

16
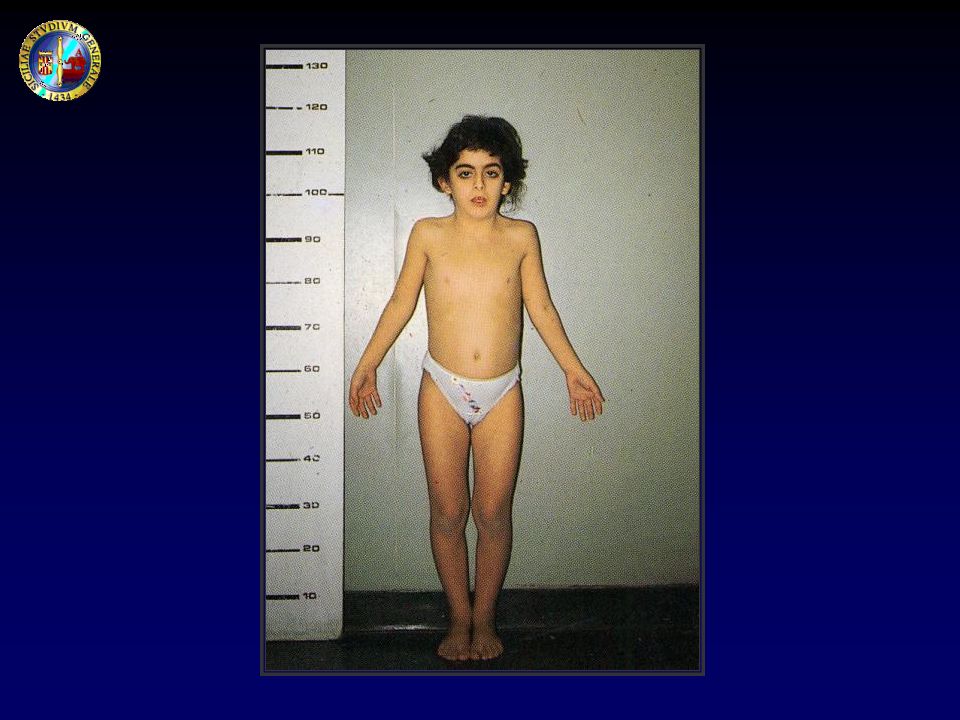
CLASSIFICAZIONE DELLE BASSE STATURE
Varianti normali dell’accrescimento Bassa statura familiare Ritardo costituzionale di crescita e pubertà Basse stature patologiche Deficit accrescitivo nelle malattie sistemiche/croniche Sindromi con o senza alterazioni cromosomiche Osteocondrodisplasie Basse stature da cause endocrine IUGR Sindrome da deprivazione affettiva

17
La statura del bambino è al di sotto della norma?
La maturazione del bambino e concorde con l’età cronologica? C’è ereditarietà per bassa statura e/o ritardo di maturazione? Quando è insorto il problema? In utero? Alla nascita? Dopo i primi anni di vita? E’ la bassa statura un sintomo isolato? La bassa statura è armonica o disarmonica?

18
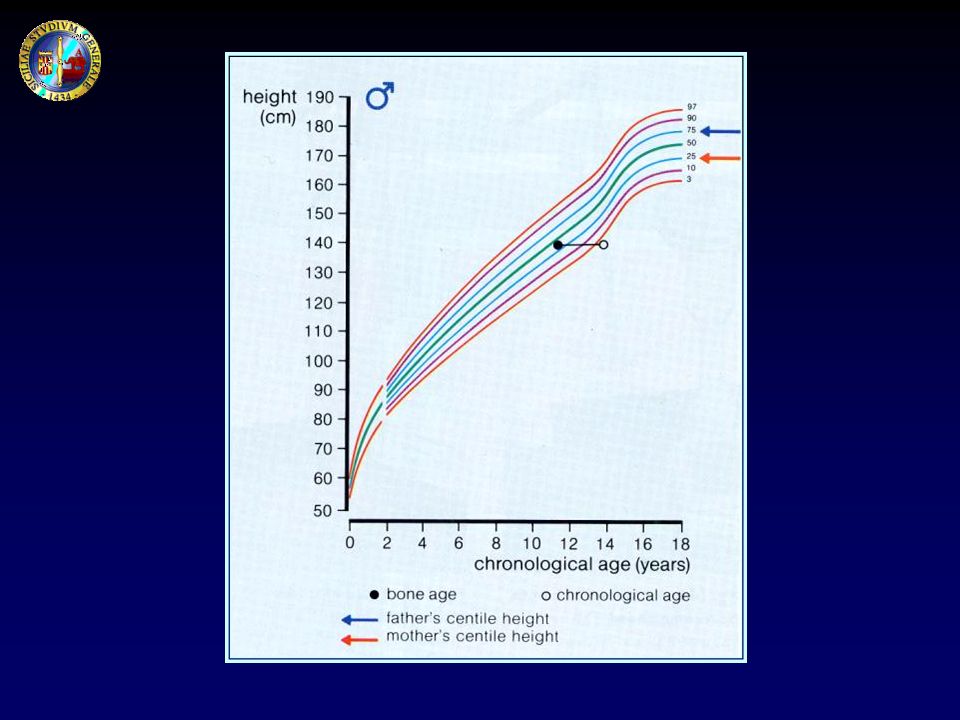
ANAMNESI Anamnesi familiare: statura, proporzioni corporee e tempo di maturazione nei genitori, fratelli, altri familiari Statura target: Statura padre + Statura madre ± 6.5 cm 2

19
Anamnesi personale: Peso, lunghezza ed età gestazionale alla nascita
Modalità del parto Segni e sintomi particolari alla nascita (linfedema al dorso dei piedi, micropene, ecc.) Precedenti diagnosi di patologie sistemiche Terapie effettuate
 Precedenti diagnosi di patologie sistemiche. Terapie effettuate.")
20
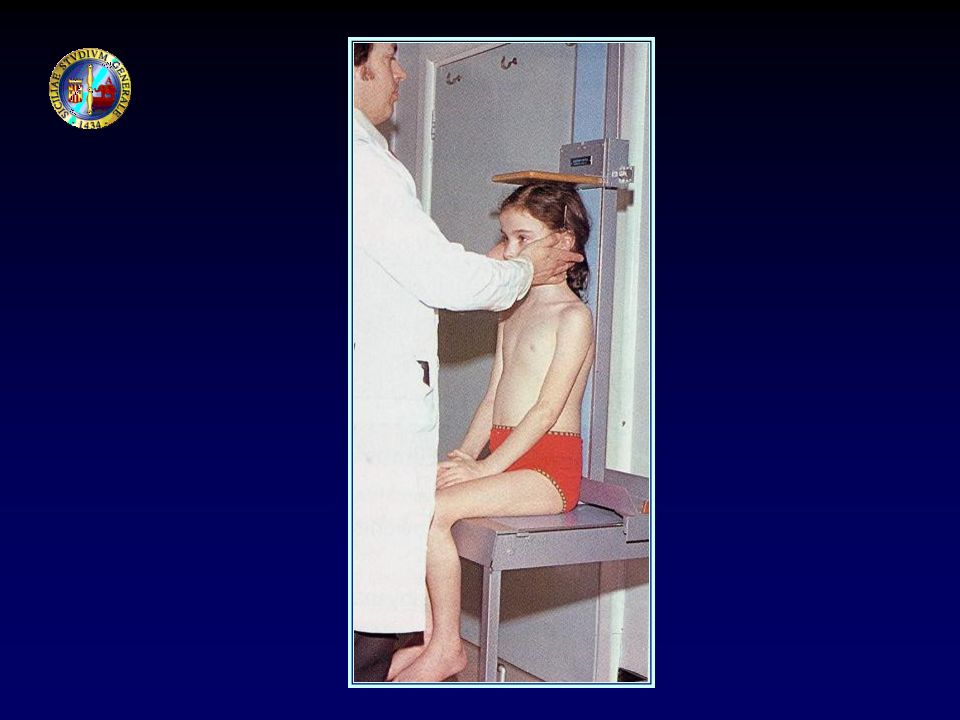
VALUTAZIONE AUXOLOGICA
Statura Peso → BMI Proporzioni corporee Statura dei genitori Maturazione ossea Stadio puberale Velocità di crescita

22
. .

24
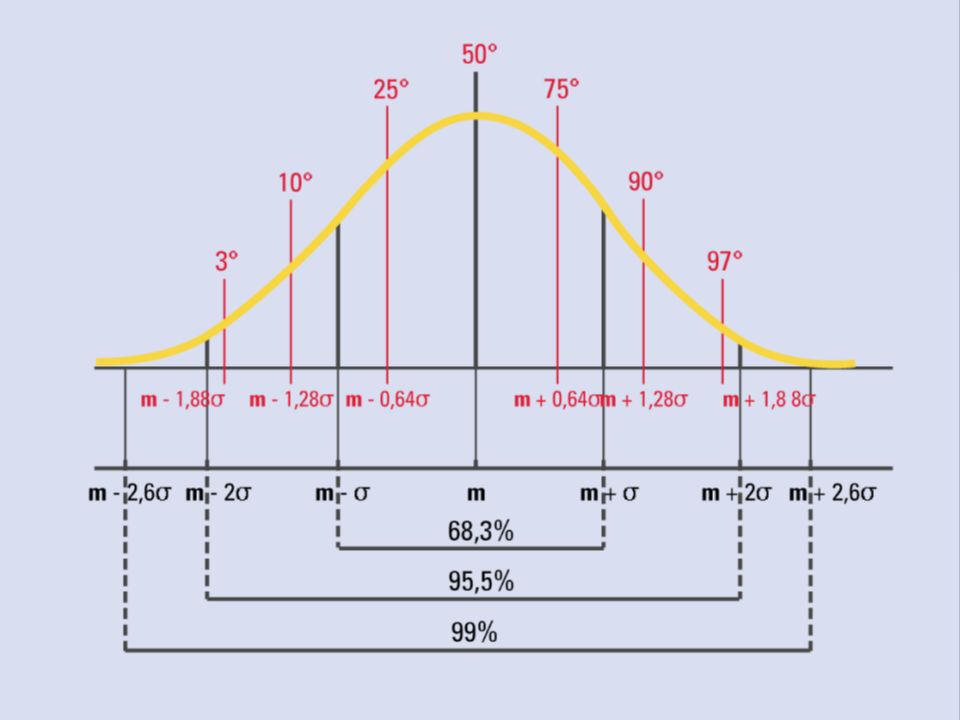
Bassa statura = statura < 3° centile o < -2 DS
Bassa statura non patologica Bassa statura patologica Velocità di crescita < 25° centile

25
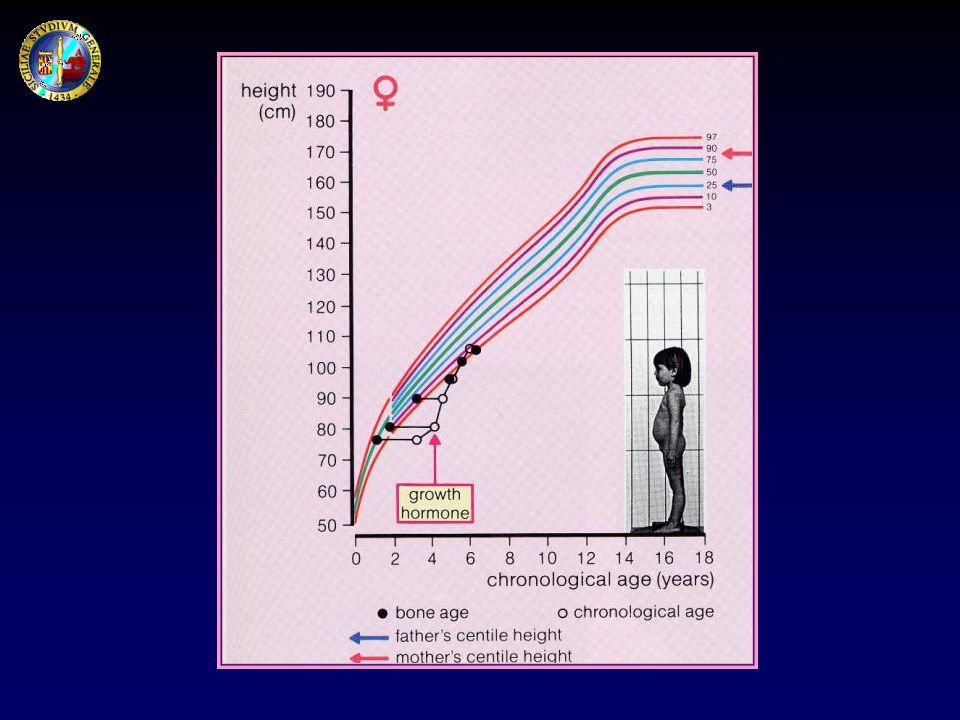
ETA’ STATURALE: corrisponde all’età che un bambino dovrebbe avere perché la sua statura venga a coincidere con la statura media per quel sesso; è molto importante rapportarla, oltre che con l’età cronologica, anche con l’età ossea

26
VALUTAZIONE DEL PESO Peso : centile, DS BMI : peso kg statura m2

27
Valutazione del peso . .

28
Valutazione del peso

29
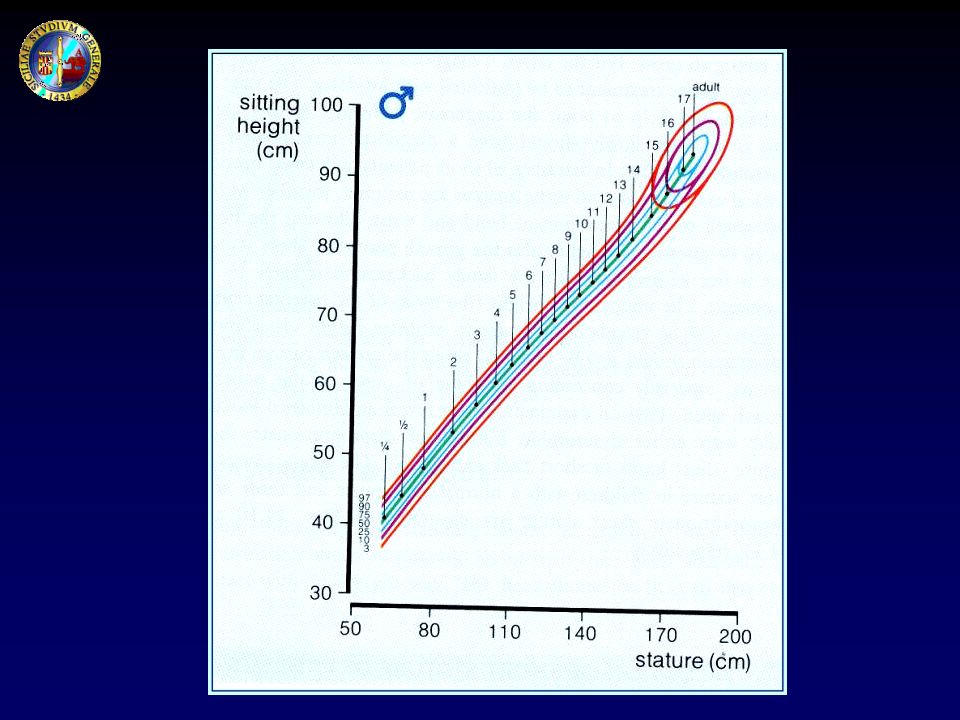
VALUTAZIONE DELLA PROPORZIONE TRA I SEGMENTI CORPOREI
Altezza da seduto Apertura braccia (arm span) Lunghezza arti inferiori
 Lunghezza arti inferiori.")
32
VALUTAZIONE DELLA MATURAZIONE OSSEA
Rx mano e polso sinistro Metodiche di valutazione : Greulich e Pyle TW2 FELS Parametro da calcolare : età ossea (percentile, DS)
")
34
UTILITA’ DELLA VALUTAZIONE DELL’ETA’ OSSEA
Diagnosi differenziale della bassa statura Predizione della statura finale Monitoraggio di un trattamento

35
INTERPRETAZIONE E SIGNIFICATO PROGNOSTICO DELL’ETA’ OSSEA
Grave ritardo: deficit ormonale di lunga durata Discreto ritardo: malattie croniche; RCCP; deprivazione affettiva Modesto ritardo: IUGR; sindromi e condizioni dismorfiche con bassa statura; displasie ossee EO/ES = 1 condizione fisiologica EO/ES < 1 prognosi staturale favorevole EO/ES > 1 prognosi staturale sfavorevole

38
VELOCITA’ DI CRESCITA OMS: “la velocità di crescita è l’indice più fedele dello stato di salute del bambino” Stagionalità Valutazione a breve o a lungo termine Relazione con lo sviluppo precoce o tardivo

40
Valutazione dei caratteri sessuali secondo Tanner
SVILUPPO PUBERALE Valutazione dei caratteri sessuali secondo Tanner Genitali Ghiandola mammaria Peluria pubica Volume testicolare Rapporto tra modificazioni della velocità di crescita e stadio puberale

41
Varianti normali della crescita
BSF RCCP Familiarità Positiva per bassa statura Positiva per ritardo EO/EC 1 < 1 EO/ES > 1 Velocità di crescita > 10°-25 ° centile > 10°-25° centile Statura bersaglio < 3° centile Normale Predizione statura adulta < 3° centile = statura bersaglio

42
BASSA STATURA FAMILIARE

43
RITARDO COSTITUZIONALE DI CRESCITA E PUBERTA’

44
INDIZI CLINICI DI BASSA STATURA PATOLOGICA
Influenze perinatali negative → Peso e lunghezza alla nascita ridotte per e.g. Patologie ereditarie → Anamnesi familiare Denutrizione / malassorbimento → Peso < statura, addome globoso Malattie croniche → Storia clinica ed esami Deprivazione psicosociale → Background sociofamiliare / disturbi del conportamento Sindromi con basse stature → Segni dismorfici, aspetto “peculiare” Osteocondrodisplasie → Alterate proporzioni corporee Disfunzioni ormonali → Rapporto peso – statura, quadro clinico ed auxologico

45
DEFICIT ACCRESCITIVO NELLE PATOLOGIE CRONICHE
Sindromi da malassorbimento → celiachia Malattie respiratorie croniche Epatopatie Insufficienza renale cronica Cardiopatie Emopatie → talassemia Fibrosi cistica Leucemie / neoplasie

46
DIAGNOSTICA DI LABORATORIO
Esami di screening Esami ormonali di 1° livello Esami ormonali di 2° livello Studio genetico

47
ESAMI DI SCREENING PER BASSA STATURA
Esame emocromocitometrico, assetto del ferro Funzionalità epatica Funzionalità renale Elettroliti sierici e urinari AGA / EMA / TTG VES, sangue occulto nelle feci FT4, TSH FSH, LH → cariotipo

49
VALUTAZIONE DELL’ASSE GH – IGF-1
Dosaggio IGF-1 (Somatomedina-C) Test di stimolo farmacologico: arginina, insulina, clonidina, L-DOPA, ecc.. Secrezione spontanea di GH nelle 12 – 24 ore
 Test di stimolo farmacologico: arginina, insulina, clonidina, L-DOPA, ecc.. Secrezione spontanea di GH nelle 12 – 24 ore.")
50
TEST DI STIMOLO FARMACOLOGICO

51
DISFUNZIONE NEUROSECRETORIA

52
DIAGNOSI STRUMENTALE Presenza di proporzioni corporee alterate: sospetta displasia ossea Rx scheletro Deficit di GH: d.d. cause deficit di GH RMN ipotalamo-ipofisi

53
STUDIO GENETICO CARIOTIPO RICERCA MUTAZIONI SPECIFICHE

54
SINDROMI CROMOSOMICHE E GENICHE CON BASSA STATURA
Progeria S. di C. De Lange S. di Bloom S. di Aarskog S. di Cockayne S. di Seckel Triploidia S. di Dubowitz S. Di Silver-Russel Trisomia 13 S. Trico-Rino-Falangea S. di Williams Trisomia 18 S. Di Smith-Lemli-Opitz S. di Noonan S.di Turner S. Di Bloom S. di Prader Willi S. di Down S.Polimalformative P. Cromosomiche

55
GENI COINVOLTI NELLE DISPLASIE OSSEE
Displasia ossea Gene mutato Acondroplasia FGFR3 Ipocondroplasia Displasia epifisaria multipla Gene per il collagene Pseudoacondroplasia COMP Discondrosteosi di Leri-Weill SHOX Pseudoipoparatiroidismo GNAS1

56
GENI COINVOLTI NELLA REGOLAZIONE DELL’ASSE DEL GH
ORGANO GENE CROMOSOMA Mutazioni identifi cate nell’uomo Ipotalamo GHRH 20 No Ipofisi GHRHR 7 Si Prop-1 5 Pit-1 3 LHX3 9 HESX1 PITX2 4 GH 17 Fegato GHR IGF-1 12 Periferia IGF-1R 15

57
BASSA STATURA DA DEFICIT DI GH
Peso e lunghezza alla nascita normali (< 50° centile se deficit congenito) Statura < 2 deviazioni standard Velocità di crescita < 10° centile EO/EC << 1 Pubertà ritardata Bassi livelli di IGF1 Risposte del GH a 2 test di stimolo farmacologico < 10 ng/ml
 Statura < 2 deviazioni standard. Velocità di crescita < 10° centile. EO/EC << 1. Pubertà ritardata. Bassi livelli di IGF1. Risposte del GH a 2 test di stimolo farmacologico < 10 ng/ml.")
58
CARATTERISTICHE CLINICHE DEL DEFICIT DI GH CONGENITO
Ipoglicemia neonatale, micropene Massiccio facciale meno sviluppato rispetto al cranio Bozze frontali Radice del naso infossata Aspetto immaturo a bambola Pannicolo adiposo abbondante soprattutto all’addome

60
RMN NEL DEFICIT DI GH

62
TERAPIA SOSTITUTIVA CON rhGH
Iniziare il più precocemente possibile Somministrazione s.c. serale in 6-7 dosi settimanali Follow-up: velocità di crescita, maturazione ossea, funzionalità tiroidea, tolleranza glucidica Va proseguita sino alla saldatura epifisaria In alcuni soggetti va proseguita anche in età adulta

63
INDICAZIONI ALL’USO DEL rhGH APPROVATE DAL MINISTERO DELLA SALUTE
Deficit di GH nel bambino Sindrome di Turner Insufficienza renale cronica Sindrome di Prader-Willi Bassa statura in bambini SGA

64
Nel corredo genetico vi è:
SINDROME DI TURNER E’ un difetto genetico che si presenta in 1 caso su nascite femminili; Nel corredo genetico vi è: solo 1 cromosoma X integro, l’altro cromosoma X è assente o parzialmente deleto Monosomia X : 45 X0 Mosaicismi: 45 X0, 46XX Anomalie della X: X ring, isocromosoma, delezione La sindrome è caratterizzata da disgenesia ovarica con conseguente infantilismo sessuale Bassa statura Segni dismorfici Malformazioni 64

65
SINDROME DI TURNER Caratteristiche fisiche della TS Statura2
Statura media adulta inferiore di 20 cm rispetto al target Linfedema a mani e piedi Accorciamento metacarpi Cubito valgo Tipiche caratteristiche facciali1,2 Ipertelorismo (allontanamento occhi) Malformazioni dell’orecchio esterno Sistema Cardiovascolare2 Anomalie valvola aortica Coartazione aortica Ipertensione Sistema Genitourinario1,2 Disgenesia Gonadica Malformazioni Renali Altre manifestazioni1,2 Nevi Diabete Iperlipidemia Tiroidite Strabismo Problemi uditivi Collo a mantello Caratteristiche fisiche della TS Lippe BM, Saenger PH. In: Sperling MA. Pediatric Endocrinology. 2nd ed. Philadelphia, PA: Saunders; 2002:519–564. Bondy CA. J Clin Endocrinol Metab 2007;92:10–25. 65 65
 Malformazioni dell’orecchio esterno. Sistema Cardiovascolare2. Anomalie valvola aortica. Coartazione aortica. Ipertensione. Sistema Genitourinario1,2. Disgenesia Gonadica. Malformazioni Renali. Altre manifestazioni1,2. Nevi. Diabete. Iperlipidemia. Tiroidite. Strabismo. Problemi uditivi. Collo a mantello. Caratteristiche fisiche della TS. Lippe BM, Saenger PH. In: Sperling MA. Pediatric Endocrinology. 2nd ed. Philadelphia, PA: Saunders; 2002:519–564. Bondy CA. J Clin Endocrinol Metab 2007;92:10–")
67
CRITERI UTILI PER LA DIAGNOSI DI S. TURNER NELLE DIVERSE ETA’
Nascita -Linfedema del dorso delle mani e dei piedi -Pterigio del collo -Anomalie cardiache -Anomalie renali Età scolare -Bassa statura -Dismorfismi Età puberale -Ritardo comparsa caratteri sessuali -Amenorrea primaria

69
Caratteristiche essenziali della TS e suo trattamento
Difetto di crescita Trattamento con GH Disgenesia gonadica e infertilità Terapia estro/progestinica ed eventualmente fertilità assistita1 Malformazioni congenite e patologie associate Approccio clinico multidisciplinare1 1. Saenger P, Wikland KA, Conway GSJ. Clin Endocrinol Metab 2001;86:3061–3069. 2. Sas TS, Mulder P, Aanstoot HJ, et al. Clin Endocrinol Metab 2001;85(2):769–775. 1. Saenger P, Wikland KA, Conway GSJ. Clin Endocrinol Metab 2001;86:3061–3069. 69
:769– Saenger P, Wikland KA, Conway GSJ. Clin Endocrinol Metab 2001;86:3061–")
70
PESO E LUNGHEZZA DEL NEONATO
Il peso alla nascita è uno dei parametri più indicativi dello stato di salute di una popolazione ed è un fattore fortemente predittivo della mortalità e morbilità neonatale Alla nascita la normalità dell’accrescimento intrauterino viene stabilita utilizzando due criteri: il peso e/o la lunghezza alla nascita e la durata della gestazione Secondo il criterio del peso avremo: neonati “di basso peso” (<2500 gr), “di peso molto basso (< 1500 gr), “ di peso bassissimo (<1000 gr) Secondo il criterio dell’età gestazionale avremo: AGA (tra -2 e 2 DS), SGA (< - 2 DS) e LGA (>2 DS) per il peso e/o la lunghezza
, di peso molto basso (< 1500 gr), di peso bassissimo (<1000 gr) Secondo il criterio dell’età gestazionale avremo: AGA (tra -2 e 2 DS), SGA (< - 2 DS) e LGA (>2 DS) per il peso e/o la lunghezza.")
71
SMALL FOR GESTATIONAL AGE (SGA) DEFINIZIONE
SGA: peso e/o lunghezza alla nascita 2 o più DS al di sotto della media per età gestazionale e sesso Circa il 5% dei nati sono SGA IUGR: documentato rallentamento della curva di crescita in utero

72
Quali sono le cause dello SGA?
Placentali • Insufficienza • Distacco • Infarto • Anormalità strutturali Materne • Infezioni • Problematiche mediche • Abuso di sostanze • Patologie della gravidanza • Età, altezza, peso alla nascita • Background etnico Fetali • Difetti cromosomici o di altra natura genetica • Malformazioni congenite • Infezioni intra-uterine • Gestazioni multiple • Sindrome di Fanconi, Bloom o Down 72

73
Peso e lunghezza di 3650 bambini nati a termine
Come riconoscerli Peso e lunghezza di 3650 bambini nati a termine 87 (2,4%) - Gruppo 3 3452 (94,6%) - Gruppo 1 54 (1,5%) - Gruppo 4 57 (1,6%) - Gruppo 2 Basso Normale Sottopeso/basso Sottopeso –2 SDS Lunghezza Peso Albertsson-Wikland K, et al Acta Paediatr, 1994; 83(Suppl 399); 64-70 73
 - Gruppo (94,6%) - Gruppo (1,5%) - Gruppo (1,6%) - Gruppo 2. Basso. Normale. Sottopeso/basso. Sottopeso. –2 SDS. Lunghezza. Peso. Albertsson-Wikland K, et al Acta Paediatr, 1994; 83(Suppl 399);")
74
CRESCITA POSTNATALE DEI BAMBINI NATI SGA
Circa il 90% presenta un “catch-up growth” (crescita di recupero) che normalizza la statura Il recupero avviene in genere entro i 2 anni 8-10% non presentano un adeguato catch-up growth Questo gruppo costituisce il 14-22% degli adulti con statura inferiore a -2 DS
 che normalizza la statura. Il recupero avviene in genere entro i 2 anni. 8-10% non presentano un adeguato catch-up growth. Questo gruppo costituisce il 14-22% degli adulti con statura inferiore a -2 DS.")
75
BASSA STATURA NEI PICCOLI PER L’ETA’ GESTAZIONALE
Peso e lunghezza alla nascita < - 2 DS Possibile catch-up growth nei primi 2 anni Scarso accrescimento ponderale EO/EC < 1 EO/ES = 1 Possibile ingresso anticipato in pubertà Ridotti livelli di IGF-1 Deficit di secrezione del GH ? Terapia con GH

76
RITARDO DI CRESCITA INTRAUTERINO

77
CRITERI PER IL TRATTAMENTO CON rhGH NEI BAMBINI SGA
Peso e/o lunghezza alla nascita < -2 DS Età superiore ai 3 anni Statura < -2.5 DS Non necessari test di stimolo del GH Dosaggio superiore a quello usato nei bambini con deficit di GH

78
TERAPIA CON rhGH IN BAMBINI SGA
Sas TC al. Clin Endocrinol 2000;53:675-81

79
ACONDROPLASIA Clinica Caratteristiche radiologiche
Bassa statura disarmonica (rizomelia) Cifoscoliosi e iperlordosi lombare Craniomegalia Ipoplasia mascellare, prognatismo mandibolare Mani a tridente Lassità ligamentosa Caratteristiche radiologiche Cranio voluminoso, base e forame magnum ridotto Riduzione della distanza interpeduncolare, canale vertebrale ristretto, corpi vertebrali corti a concavità posteriore Bacino quadrangolare, forami ischiatici piccoli e tetti acetabolari orizzontali Colli femorali corti, tozzi Ossa lunghe brevi, metafisi slargate
 Cifoscoliosi e iperlordosi lombare. Craniomegalia. Ipoplasia mascellare, prognatismo mandibolare. Mani a tridente. Lassità ligamentosa. Caratteristiche radiologiche. Cranio voluminoso, base e forame magnum ridotto. Riduzione della distanza interpeduncolare, canale vertebrale ristretto, corpi vertebrali corti a concavità posteriore. Bacino quadrangolare, forami ischiatici piccoli e tetti acetabolari orizzontali. Colli femorali corti, tozzi. Ossa lunghe brevi, metafisi slargate.")
80
ACONDROPLASIA Complicanze Terapia
Neurologiche (idrocefalo, compressione midollare, ipotonia) Respiratorie ( apnee ostruttive e centrali, torace ristretto) Otorinolaringoiatriche (otiti, ipoacusia, infezioni vie respiratorie) Obesità Odontoiatriche (prognatismo, malocclusione) Terapia GH (risposta per lo più insoddisfacente) Allungamento chirurgico degli arti Ortodonzia Dieta
 Respiratorie ( apnee ostruttive e centrali, torace ristretto) Otorinolaringoiatriche (otiti, ipoacusia, infezioni vie respiratorie) Obesità. Odontoiatriche (prognatismo, malocclusione) Terapia. GH (risposta per lo più insoddisfacente) Allungamento chirurgico degli arti. Ortodonzia. Dieta.")
81
IPOCONDROPLASIA Caratteristiche radiologiche
Riduzione della distanza interpeduncolare L1-L5 Conformazione alterata delle vertebre lombari Ali iliache squadrate Colli femorali corti, tozzi Fibula più lunga della tibia Brachidattilia

82
IL GENE SHOX SHOX è situato nelle regioni pseudoautosomiche (PAR1) dei cromosomi sessuali X e Y I geni della regione PAR1 sfuggono all’inattivazione pertanto due copie attive del gene sono necessarie per il suo normale funzionamento Mutazioni, delezioni, assenza di una copia (aploinsufficienza) o di entrambe le copie del gene sono associati a bassa statura e anomalie scheletriche APLOINSUFFICIENZA Sindrome di Turner (TS) Sindrome di Leri-Weill o Discondrosteosi di Leri- Weill (LWD) PERDITA OMOZIGOTE Sindrome di Langer o Displasia Mesomelica di Langer (MDL)
 o di entrambe le copie del gene sono associati a bassa statura e anomalie scheletriche. APLOINSUFFICIENZA. Sindrome di Turner (TS) Sindrome di Leri-Weill o Discondrosteosi. di Leri- Weill (LWD) PERDITA OMOZIGOTE. Sindrome di Langer o Displasia. Mesomelica di Langer (MDL)")
83
La Sindrome di Léri-Weill (LWS) o discondrosteosi di Léri-Weill
Il quadro fenotipico completo è caratterizzato da: Bassa statura Sproporzione degli arti con mesomelia Deformità di Madelung Parziale dislocazione dell’ulna nel polso, nel gomito o in entrambi Incuneamento delle ossa del carpo tra radio e ulna Movimento limitato di polso e/o gomito possono associarsi: 4° metacarpo/metatarso brevi Curvatura di radio/ulna/tibia Esostosi di tibia/perone Anormalità del collo femorale/tuberosità dell’omero Ipertrofia muscolare (polpacci)
")
84
LWD Ross JL J Pediatr 2005, 147,499 Binder G. JCEM 88; 4891,2003 84

85
Il nanismo mesomelico o displasia mesomelica di Langer
Il quadro fenotipico è caratterizzato da: deviazione ulnare delle mani assottigliamento distale dell’omero e della fibula ipoplasia del radio e dell’ulna bassa statura mesomelica grave, con arti ipoplasici o corti, che possono presentare anche malformazioni o saldature anomale ipoplasia della mandibola

86
Ritardo costituzionale 25% Bassa statura familiare 11% RC + BS 4%
INCIDENZA DELLE VARIE CAUSE DI BASSA STATURA SU 748 PAZIENTI (DA BRASEL E BLIZZARD) Ritardo costituzionale 25% Bassa statura familiare 11% RC + BS 4% Deficit di GH idiopatico 4% Deficit di GH organica 3% Disgenesia gonadica 7% Nanismo psicosociale 6% Condrodisplasia % Ipotiroidismo primario 2% IUGR 4% Morbo di Crohn ,4% Altre cause %
 Ritardo costituzionale 25% Bassa statura familiare 11% RC + BS 4% Deficit di GH idiopatico 4% Deficit di GH organica 3% Disgenesia gonadica 7% Nanismo psicosociale 6% Condrodisplasia 3% Ipotiroidismo primario 2% IUGR 4% Morbo di Crohn 1,4% Altre cause 25%")








 formazione del centro di ossificazione b) accrescimento c) modellamento e mineralizzazione Es.: ossa del cranio,>")
















 in un individuo adulto in grado di procreare.>")
>")