Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Predizione della struttura di una proteina
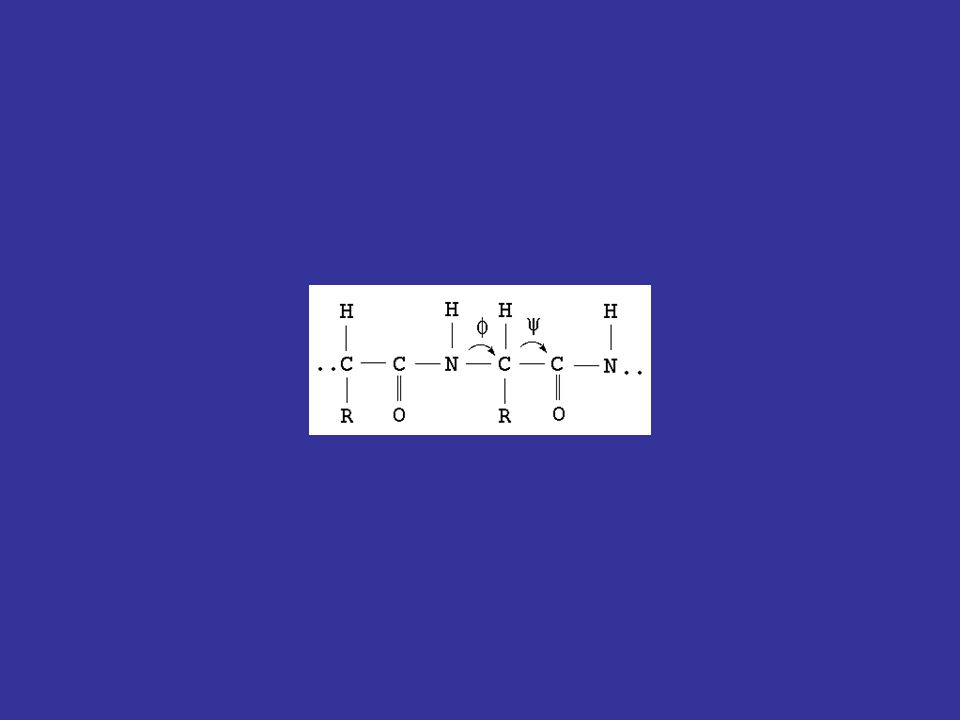
In generale, la sequenza amminoacidica di una proteina codifica la sua struttura tridimensionale dovrebbe quindi essere possibile disegnare un algoritmo per predire la struttura 3D di una proteina a partire dalla sua sequenza 1D questo è l’obiettivo più ambizioso e complesso della bioinformatica, e non è ancora stato raggiunto

2
Swiss-Prot + TrEMBL 700.000 sequenze
i metodi sperimentali per la determinazione della sequenza di una proteina sono estremamente rapidi (l’ordine di grandezza è il giorno) e relativamente economici la risoluzione della struttura tridimensionale di una proteina richiede invece l’uso di strumenti più complessi, e talvolta mesi di lavoro Swiss-Prot + TrEMBL sequenze PDB (Protein Data Bank) strutture gran parte delle ricerche in biologia strutturale è quindi volta allo studio delle leggi fondamentali del folding delle proteine e la biologia computazionale dedica molte energie e risorse allo sviluppo di metodi per la predizione della struttura delle proteine
 e relativamente economici. la risoluzione della struttura tridimensionale di una proteina richiede invece l’uso di strumenti più complessi, e talvolta mesi di lavoro. Swiss-Prot + TrEMBL sequenze. PDB (Protein Data Bank) strutture. gran parte delle ricerche in biologia strutturale è quindi volta allo studio delle leggi fondamentali del folding delle proteine e la biologia computazionale dedica molte energie e risorse allo sviluppo di metodi per la predizione della struttura delle proteine.")
3
la contiguità dei residui in struttura determina la funzione
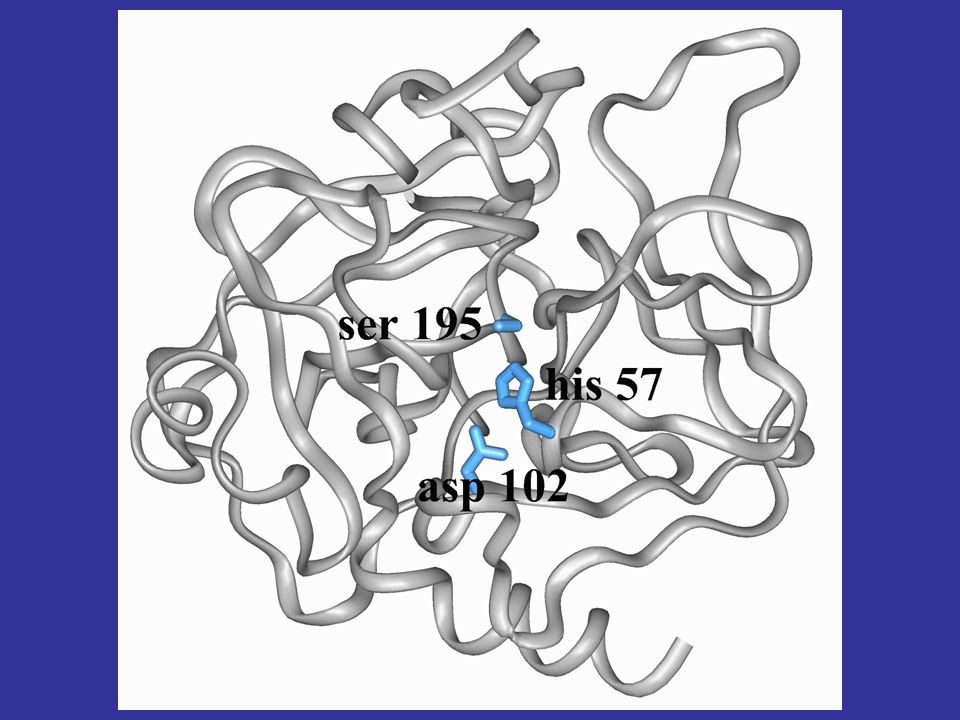
perchè è interessante studiare la struttura di una proteina in molti casi è vero che solo l’analisi della struttura tridimensionale di una macromolecola può aiutarci a comprendere in quale modo e per quale motivo una determinata sequenza (avvolta in una specifica struttura) possa codificare una ben precisa funzione vediamo per esempio la struttura 3D della chimotripsina, mettendo in evidenza i residui della triade catalitica, che non sono contigui nella sequenza proteica la contiguità dei residui in struttura determina la funzione
 possa codificare una ben precisa funzione. vediamo per esempio la struttura 3D della chimotripsina, mettendo in evidenza i residui della triade catalitica, che non sono contigui nella sequenza proteica. la contiguità dei residui in struttura determina la funzione.")
6
RNase A (A) RNase T1 (B) Wheatwin1 (C)
 RNase T1 (B) Wheatwin1 (C)")
7
Metodi per la predizione della struttura secondaria
Il metodo Chou-Fasman

8
a elica foglietto b ripiegamento b aa C&F L pr Glu 1.51 1.44 ++ Val 1.70 1.49 Asn 1.56 1.28 Met 1.45 1.47 Ile 1.60 Gly 1.64 Ala 1.42 1.29 Tyr 1.25 Pro 1.52 1.91 Leu 1.21 1.30 Phe 1.38 1.32 + Asp 1.46 1.41 Lys 1.16 1.23 Trp 1.37 1.14 Ser 1.43 1.13 1.07 1.02 Cys 1.19 0.81 Gln 1.11 1.27 0.74 1.05 1.08 0.99 Thr 1.01 0.96 0.97 1.10 0.80 0.98 1.06 0.91 1.04 = Arg 0.93 0.76 His 1.00 1.22 0.89 0.95 0.88 0.87 0.68 0.83 0.82 0.90 - 0.77 0.75 0.66 0.70 0.92 0.60 0.41 0.69 0.72 0.59 0.67 0.55 0.64 -- 0.58 0.57 0.52 0.54 0.50 0.47 0.56 0.37 0.51 Tabella 5.1. Propensione dei residui aminoacidici a formare elementi di struttura secondaria come riportato da Chou-Fasman (1978b) (C&F) e Levitt (1978) (L). La colonna “pr” classifica i residui come indifferenti (=) o stabilizzatori/destabilizzatori forti (++/--) e deboli (+/-) della struttura secondaria.
 (C&F) e Levitt (1978) (L). La colonna pr classifica i residui come indifferenti (=) o stabilizzatori/destabilizzatori forti (++/--) e deboli (+/-) della struttura secondaria.")
9
Metodi di predizione della struttura secondaria delle proteine:
Metodi di Chou-Fasman si basa sull’analisi statistica della composizione in residui delle strutture secondarie presenti nella PDB. ( GOR si basa sull’analisi statistica della composizione in residui delle strutture secondarie presenti nella PDB. ( AGADIR per predire la percentuale di residui in elica ( PHD prende in input o una sequenza o un allineamento multiplo ed usa le reti neurali. ( Vuole una registrazione PSIPRED utilizza un sistema di due reti neurali. ( PREDATOR si basa sull’applicazione del metodo del k-esimo vicino che usa le reti neurali ( JPRED3 ( fa un consensus di vari metodi
 GOR si basa sull’analisi statistica della composizione in residui delle strutture secondarie presenti nella PDB. ( page=npsa_gor4.html) AGADIR per predire la percentuale di residui in elica. ( PHD prende in input o una sequenza o un allineamento multiplo ed usa le reti neurali. ( Vuole una registrazione. PSIPRED utilizza un sistema di due reti neurali. ( PREDATOR si basa sull’applicazione del metodo del k-esimo vicino che usa le reti neurali. ( JPRED3 ( fa un consensus di vari metodi.")
10
I migliori programmi di predizione della struttura secondaria sono stati sviluppati utilizzando metodi di apprendimento automatico (machine learning methods) i metodi di apprendimento automatico più utilizzati in bioinformatica sono le reti neurali e gli Hidden Markov Models (HMM) una caratteristica peculiare delle reti neurali è che sono in grado di apprendere, in un tentativo di simulare il comportamento del cervello umano vengono addestrate utilizzando un opportuno insieme di dati detto training set (un insieme di a-eliche, filamenti b e elementi non-a non-b) e possono poi venire utilizzate per riconoscere a-eliche da filamenti b e da elementi non-a non-b
 una caratteristica peculiare delle reti neurali è che sono in grado di apprendere, in un tentativo di simulare il comportamento del cervello umano. vengono addestrate utilizzando un opportuno insieme di dati detto training set (un insieme di a-eliche, filamenti b e elementi non-a non-b) e possono poi venire utilizzate per riconoscere a-eliche da filamenti b e da elementi non-a non-b.")
13
output di PredictProtein (PHD)
un programma di predizione della struttura secondaria e accessibilità al solvente ...., , , , , ,....6 AA |MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAG| PHD sec | EEEEEEEE HHHHHHHHHHH EEEEE HHHHHHH | Rel sec | | detail: prH sec | | prE sec | | prL sec | | subset: SUB sec |L.EEEEEEE.LLLLLHHHHHHHHHHH.....LLLL.....L.EEE..LL...HHHH..LL| ...., , , , , ,....12 AA |QEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDL| PHD sec | HHHHHHHHHHHHH EEEEEE HHHHHHHHHHHHHHH EEEEEE HH| Rel sec | | prH sec | | prE sec | | prL sec | | subset: SUB sec |..HHHHHHHHHHH..LL.EEEEEE.LLLL.HHHHHHHHHHHHH..LLLL.EEEE.LL..H|

14
riconoscimento del fold (fold recognition)
banca dati di fold possibili sequenza query input allineamento sequenza struttura + output

15
foglietto -elica filamento

16
perchè è interessante studiare la struttura di una proteina
il confronto tra sequenze proteiche può essere utilizzato per mettere in luce relazioni evolutive tra proteine e la similarità tra sequenze può essere utilizzata come una misura della distanza evolutiva tra gli organismi come abbiamo visto, le proteine che si confrontano possono talvolta essere così diverse che diventa difficile metterne in evidenza la comune origine evolutiva attraverso il solo confronto tra sequenze cambiamenti nella struttura delle proteine sono invece più conservativi: l’evoluzione delle proteine avviene in modi che in genere non alterano il ripiegamento (fold) della struttura proteica, che quindi può conservare tracce di un’origine comune
 della struttura proteica, che quindi può conservare tracce di un’origine comune.")
17
la cristallografia a raggi X
Come si può studiare la struttura di una proteina i metodi sperimentali classici per la risoluzione della struttura tridimensionale di una proteina sono: la cristallografia a raggi X la spettroscopia a risonanza magnetica e nucleare (Nuclear Magnetic Resonance, NMR)
")
18
cellula batterica moltiplicazione del clone purificazione
diffrazione ai raggi X cellula batterica plasmide DNA esogeno formazione di cristalli NMR moltiplicazione del clone purificazione della proteina

19
x y z nome composto organismo autore referenze risoluzione sequenza
HEADER TRANSCRIPTION REGULATION AUG RPO RPO 2 COMPND ROP (COLE1 REPRESSOR OF PRIMER) MUTANT WITH ALA INSERTED ON 1RPO 3 COMPND 2 EITHER SIDE OF ASP 31 (INS (A-D31-A)) RPO 4 SOURCE (ESCHERICHIA COLI) RPO 5 AUTHOR M.VLASSI,M.KOKKINIDIS RPO 6 REVDAT MAY-95 1RPOA REMARK RPOA 1 REVDAT FEB-95 1RPO RPO 7 JRNL AUTH M.VLASSI,C.STEIF,P.WEBER,D.TSERNOGLOU,K.WILSON, RPO 8 JRNL AUTH 2 H.J.HINZ,M.KOKKINIDIS RPO 9 JRNL TITL RESTORED HEPTAD PATTERN CONTINUITY DOES NOT RPO 10 JRNL TITL 2 ALTER THE FOLDING OF A 4-ALPHA-HELICAL BUNDLE RPO 11 JRNL REF NAT.STRUCT.BIOL V RPO 12 JRNL REFN ASTM NSBIEW US ISSN RPO 13 REMARK RPO 14 REMARK 1 REFERENCE RPO 15 REMARK 1 AUTH M.KOKKINIDIS,M.VLASSI,Y.PAPANIKOLAOU,D.KOTSIFAKI, 1RPO 16 REMARK 1 AUTH 2 A.KINGSWELL,D.TSERNOGLOU,H.J.HINZ RPO 17 REMARK 1 TITL CORRELATION BETWEEN PROTEIN STABILITY AND CRYSTAL 1RPO 18 REMARK 1 TITL 2 PROPERTIES OF DESIGNED ROP VARIANTS RPO 19 REMARK 1 REF PROTEINS.STRUCT.,FUNCT., V RPOA 2 REMARK 1 REF 2 GENET RPOA 3 REMARK 1 REFN ASTM PSFGEY US ISSN RPO 22 REMARK RPO 29 REMARK 2 RESOLUTION ANGSTROMS RPO 30 REMARK RPO 94 REMARK 999 SEQUENCE NUMBER IS ALSO THAT FROM PDB ENTRY RPO 95 SEQRES MET THR LYS GLN GLU LYS THR ALA LEU ASN MET ALA ARG 1RPO 96 SEQRES PHE ILE ARG SER GLN THR LEU THR LEU LEU GLU LYS LEU 1RPO 97 SEQRES ASN GLU LEU ALA ASP ALA ALA ASP GLU GLN ALA ASP ILE 1RPO 98 SEQRES CYS GLU SER LEU HIS ASP HIS ALA ASP GLU LEU TYR ARG 1RPO 99 SEQRES SER CYS LEU ALA ARG PHE GLY ASP ASP GLY GLU ASN LEU 1RPO 100 ATOM N MET RPO 115 ATOM CA MET RPO 116 ATOM C MET RPO 117 ATOM O MET RPO 118 ATOM CB MET RPO 119 ATOM CG MET RPO 120 ATOM SD MET RPO 121 ATOM CE MET RPO 122 ATOM N THR RPO 123 ATOM CA THR RPO 124 ATOM C THR RPO 125 autore organismo referenze risoluzione sequenza residuo 1 residuo 2 num. residuo tipo residuo num.atomo tipo atomo x y z
 MUTANT WITH ALA INSERTED ON 1RPO 3. COMPND 2 EITHER SIDE OF ASP 31 (INS (A-D31-A)) 1RPO 4. SOURCE (ESCHERICHIA COLI) 1RPO 5. AUTHOR M.VLASSI,M.KOKKINIDIS 1RPO 6. REVDAT 2 15-MAY-95 1RPOA 1 REMARK 1RPOA 1. REVDAT 1 14-FEB-95 1RPO 0 1RPO 7. JRNL AUTH M.VLASSI,C.STEIF,P.WEBER,D.TSERNOGLOU,K.WILSON, 1RPO 8. JRNL AUTH 2 H.J.HINZ,M.KOKKINIDIS 1RPO 9. JRNL TITL RESTORED HEPTAD PATTERN CONTINUITY DOES NOT 1RPO 10. JRNL TITL 2 ALTER THE FOLDING OF A 4-ALPHA-HELICAL BUNDLE 1RPO 11. JRNL REF NAT.STRUCT.BIOL. V RPO 12. JRNL REFN ASTM NSBIEW US ISSN RPO 13. REMARK 1 1RPO 14. REMARK 1 REFERENCE 1 1RPO 15. REMARK 1 AUTH M.KOKKINIDIS,M.VLASSI,Y.PAPANIKOLAOU,D.KOTSIFAKI, 1RPO 16. REMARK 1 AUTH 2 A.KINGSWELL,D.TSERNOGLOU,H.J.HINZ 1RPO 17. REMARK 1 TITL CORRELATION BETWEEN PROTEIN STABILITY AND CRYSTAL 1RPO 18. REMARK 1 TITL 2 PROPERTIES OF DESIGNED ROP VARIANTS 1RPO 19. REMARK 1 REF PROTEINS.STRUCT.,FUNCT., V RPOA 2. REMARK 1 REF 2 GENET. 1RPOA 3. REMARK 1 REFN ASTM PSFGEY US ISSN RPO 22. REMARK 2 1RPO 29. REMARK 2 RESOLUTION. 1.4 ANGSTROMS. 1RPO 30. REMARK 1RPO 94. REMARK 999 SEQUENCE NUMBER IS ALSO THAT FROM PDB ENTRY 1RPO 95. SEQRES 1 65 MET THR LYS GLN GLU LYS THR ALA LEU ASN MET ALA ARG 1RPO 96. SEQRES 2 65 PHE ILE ARG SER GLN THR LEU THR LEU LEU GLU LYS LEU 1RPO 97. SEQRES 3 65 ASN GLU LEU ALA ASP ALA ALA ASP GLU GLN ALA ASP ILE 1RPO 98. SEQRES 4 65 CYS GLU SER LEU HIS ASP HIS ALA ASP GLU LEU TYR ARG 1RPO 99. SEQRES 5 65 SER CYS LEU ALA ARG PHE GLY ASP ASP GLY GLU ASN LEU 1RPO 100. ATOM 1 N MET RPO 115. ATOM 2 CA MET RPO 116. ATOM 3 C MET RPO 117. ATOM 4 O MET RPO 118. ATOM 5 CB MET RPO 119. ATOM 6 CG MET RPO 120. ATOM 7 SD MET RPO 121. ATOM 8 CE MET RPO 122. ATOM 9 N THR RPO 123. ATOM 10 CA THR RPO 124. ATOM 11 C THR RPO 125. autore. organismo. referenze. risoluzione. sequenza. residuo 1. residuo 2. num. residuo. tipo residuo. num.atomo. tipo atomo. x. y. z.")
21
myoglobin AND aplysia parole-chiave

24
utilizzo di un semplice programma di grafica molecolare RasMol

26
Metodi computazionali per la predizione della struttura tridimensionale proteica:
quali sono e che grado di affidabilità offrono? i metodi ab initio sono i più ambiziosi, ma i tempi di calcolo sono ancora TROPPO LUNGHI perché possano anche avere un interesse pratico i metodi knowledge based sono attualmente quelli che sembrano funzionare meglio, anche se sono meno soddisfacenti dal punto di vista della comprensione ultima dei meccanismi naturali del folding delle proteine

27
obiettivi intermedi e meno ambiziosi
la predizione della struttura secondaria: quali segmenti della sequenza formano a-eliche e quali filamenti b o anse? il riconoscimento del fold (fold recognition o threading): data una sequenza proteica e un insieme di possibili fold tridimensionali, è possibile identificare il fold più simile a quello davvero assunto dalla sequenza? il modelling per omologia (homology modelling): se una proteina a struttura non nota è omologa ad una proteina a struttura nota, possiamo assumere che le due strutture siano simili e usare l’una per generare un modello dell’altra?
: data una sequenza proteica e un insieme di possibili fold tridimensionali, è possibile identificare il fold più simile a quello davvero assunto dalla sequenza il modelling per omologia (homology modelling): se una proteina a struttura non nota è omologa ad una proteina a struttura nota, possiamo assumere che le due strutture siano simili e usare l’una per generare un modello dell’altra")
28
modelling per omologia (homology modelling)
La qualità del modello dipende dalla similarità tra le sequenze delle due proteine in generale, a maggiore identità di sequenza tra due proteine, corrisponde maggiore similarità tra le corrispondenti strutture se l’identità tra due sequenze proteiche è superiore al 30%, si può assumere che le loro strutture saranno simili se una proteina a struttura non nota ha almeno il 30% di identità con una proteina a struttura nota, si può usare la seconda come templato per generare un modello per omologia della prima

29
Come nel confronto di sequenze e’ necessario allinearle, nel confronto di strutture 3D e’ necessario sovrapporle come corpi rigidi scegliendo una regola di corrispondenza tra coppie di atomi o di residui nelle due strutture. La prima difficolta’ consiste nel fatto che le due proteine molto spesso non hanno lo stesso numero di residui. Per la sovrapposizione si possono utilizzare le catene dei carboni alfa appartenenti agli elementi di struttura secondaria perche’ in genere le inserzioni e delezioni si accumulano nei loops che possono semplicemente venire esclusi dalla sovrapposizione. I metodi di confronto 3D utilizzano l’ allineamento delle sequenze per decidere la regola di corrispondenza alla base della sovrapposizione strutturale

30
Un allineamento strutturale può essere valutato in base alla deviazione quadratica media (root mean square deviation o r.m.s.d.), al numero di atomi che sono stati accoppiati nella sovrapposizione e alla valutazione della similarità dei residui sovrapposti. L’r.m.s.d. o r.m.s. di una sovrapposizione tridimensionale è la distanza media tra gli atomi di tutte le coppie che hanno partecipato all’allineamento strutturale, per cui tanto più bassa è l’r.m.s. tanto migliore sarà l’allineamento strutturale calcolato D = distanza tra coppie di atomi appaiati N = numero di coppie considerate

31
valutazione dell’allineamento strutturale
un altro criterio di valutazione di un allineamento strutturale è rappresentato dal numero di atomi o di residui che sono stati accoppiati si cerca di massimizzare il numero di atomi accoppiati e di minimizzare la corrispondente r.m.s. a parità di numero di residui accoppiati, il migliore allineamento strutturale sarà quello con minore r.m.s. a parità di r.m.s. verrà considerato migliore l’allineamento strutturale operato con un maggior numero di atomi accoppiati oltre a questi due valori tipici delle sovrapposizioni tridimensionali, si può anche considerare il punteggio di similarità dei residui accoppiati

32
Grafico identità di sequenza/rms tra le strutture
2,5 2,0 1,5 r.m.s.d.tra atomi della catena principale del core 1,0 0,5 0,0 100 75 50 25 % di residui identici nel core proteico

33
Lisozima di pollo Alpha-lactalbumina di babbuino 37% identità
1 KQFTKCELSQ NLYD--IDGY GRIALPELIC TMFHTSGYDT QAIVENDE-S TEYGLFQISN ALWCKSSQSP QSRNICDITC DKFLDDDITD DIMCAKKILD 1 KVFGRCELAA AMKRHGLDNY RGYSLGNWVC AAKFESNFNT QATNRNTDGS TDYGILQINS RWWCNDGRTP GSRNLCNIPC SALLSSDITA SVNCAKKIVS * * .*** * * .* . .* . * ..* ** * . * *.**..**.. **. ...* ***.*.* * .* *** . *****. 98 IK-GIDYWIA HKALCT-EKL EQWL--CEK- 101 DGNGMNAWVA WRNRCKGTDV QAWIRGCRL *.. *.* . * *. * 37% identità di sequenza

34
Predizione della struttura terziaria - diagramma di flusso
Confronto con banche dati di sequenze proteiche Allineamento di sequenze. E’ nota la struttura? sì no Predizione di struttura secondaria Ricerche di motivi, fold recognition, ab initio Modelling per omologia usando coordinate di proteina a struttura nota sì Valutazione accuratezza della predizione

35
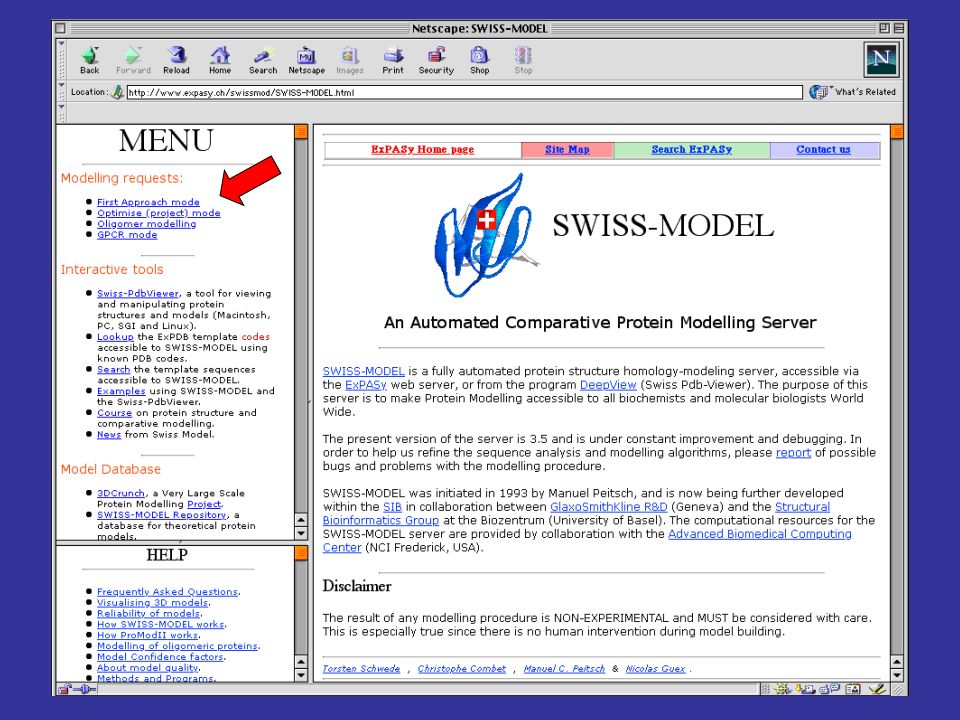
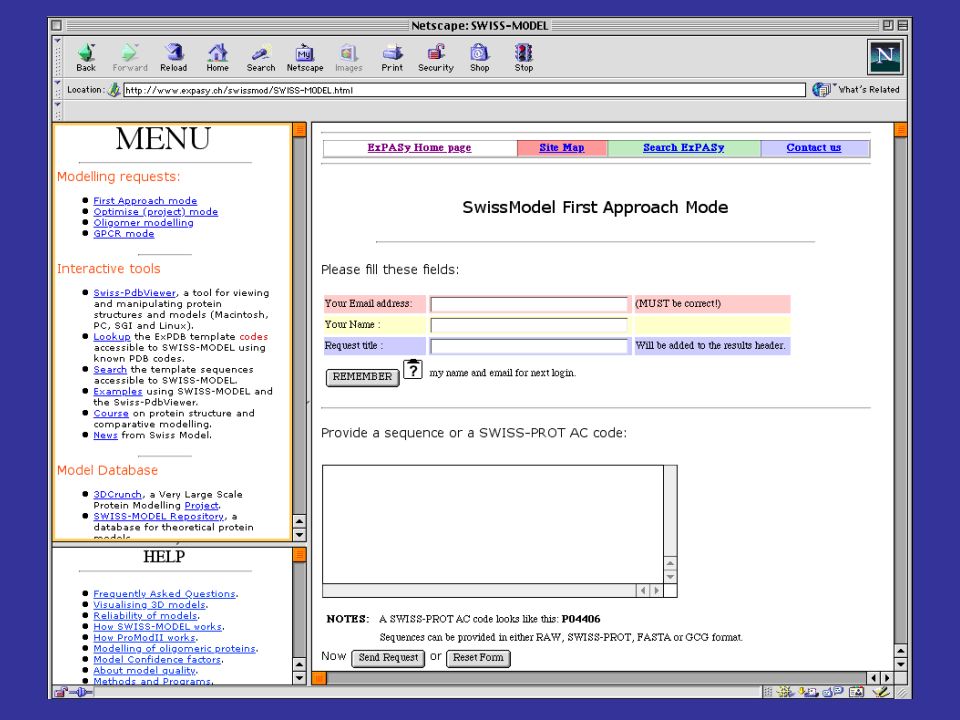
per costruire modelli per omologia di proteine che abbiamo una alta identità di sequenza con una proteina a struttura nota, si può utilizzare il server automatico SwissModel SwissModel è disponibile all’indirizzo: prende in input una sequenza proteica e l’indirizzo di posta elettronica della persona che sottomette la sequenza cerca nel PDB possibili strutture che possano fare da templato (che abbiano cioè una buona identità di sequenza con la proteina query) spedisce con la posta elettronica le coordinate del modello o le motivazioni della propria incapacità a produrne uno di buona affidabilità
 spedisce con la posta elettronica le coordinate del modello o le motivazioni della propria incapacità a produrne uno di buona affidabilità.")
Presentazioni simili







>")
>")






![PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]](/1/540371/big_thumb.jpg "PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]>")






