Scaricare la presentazione
1
I programmi di ricerca in banche dati possono essere oppure essere utilizzabili via web residenti in un calcolatore di cui siamo proprietari o utenti

2
Il concetto di directory directory principale primo livello di sub-directories secondo livello di sub-directories bio1 acidi nucleici proteine coli mouse human

3
Come si effettua una ricerca in una banca dati si possono effettuare ricerche utilizzando parole-chiave (es.: emoglobina) e ricavando i nomi dei files che le contengono oppure si possono utilizzare sequenze in input per ricavare liste di sequenze simili ad esse
 e ricavando i nomi dei files che le contengono oppure si possono utilizzare sequenze in input per ricavare liste di sequenze simili ad esse")
4
a cosa serve effettuare ricerche per similarità di sequenza in banche dati confronto tra sequenze costruzione di alberi filogenetici identificazione di domini funzionali costruzione di modelli per omologia in 3D

5
ATTENZIONE alla proprietà di linguaggio! due sequenze si dicono omologhe se condividono una stessa origine filogenetica é un dato che prescinde da eventuali ipotesi sulla causa della similarità stessa similaritàomologia

6
La similarità biologica è spesso dovuta ad omologia, ma può anche presentarsi per caso oppure per fenomeni di convergenza adattativa Ad esempio: lala di un uccello e lala di un pipistrello si sono evolute indipendentemente e di conseguenza non sono omologhe Nel trattare le sequenze è sempre più corretto utilizzare il termine similarità, in quanto è sempre possibile stabilire quanto due sequenze siano simili, mentre non sempre si può decidere se la similarità sia dovuta ad omologia, a convergenza adattativa, oppure al caso

7
un po di terminologia… strutture o sequenze ortologhe in due organismi sono sequenze omologhe che sono evolute dalla stessa caratteristica nel loro ultimo antenato comune ma che non necessariamente mantengono la loro funzione ancestrale. sequenze omologhe la cui evoluzione riflette invece eventi di duplicazione genica si definiscono paraloghe. per esempio, la catena dell emoglobina e un paralogo della catena dell emoglobina e della mioglobina, dal momento che ambedue si sono evolute dallo stesso gene ancestrale attraverso ripetuti eventi di duplicazione genica.

8
in questa prima parte della lezione ci occuperemo solo di COPPIE di sequenze

9
calcolo della similarità tra due sequenze date similarità allineamento non si possono allineare due sequenze senza definire criteri si similarità per valutare la similarità tra due sequenze, dobbiamo prima allinearle

10
INPUT acidi nucleici (4 possibili residui nucleotidici) proteine (20 possibili residui aminoacidici) la bioinformatica in generale studia le sequenze come stringhe di caratteri calcolo della similarità tra due sequenze date
 proteine (20 possibili residui aminoacidici) la bioinformatica in generale studia le sequenze come stringhe di caratteri calcolo della similarità tra due sequenze date")
11
cominciamo quindi col definire una prima semplice misura di similarità, data dalla somma dei caratteri delle due sequenze che si appaiano esattamente calcolo della similarità tra due sequenze date facciamo scorrere una delle due sequenze sullaltra in tutte le posizioni possibili (generiamo tutti i possibili allineamenti) e valutiamo la similarità di sequenza di ognuno degli allineamenti generati
 e valutiamo la similarità di sequenza di ognuno degli allineamenti generati")
12
calcolo della similarità tra due sequenze date lallineamento associato alla più alta valutazione della similarità di sequenza verrà scelto come il migliore degli allineamenti possibili esempio: AAKKQW AAKQW definiamo similarità di sequenza tra le due sequenze come il più alto dei punteggi ottenuti

13
AAKKQW AAKQW calcolo della similarità tra due sequenze date (senza gaps) AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW 0 0 0 0 4 3 1 0 0 0
 AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW")
14
calcolo della similarità tra due sequenze date (senza gaps) AAKQW AAKKQW 6 caratteri 5 caratteri abbiamo valutato 10 (5+5) allineamenti abbiamo confrontato 30 (6x5) caratteri
 AAKQW AAKKQW 6 caratteri 5 caratteri abbiamo valutato 10 (5+5) allineamenti abbiamo confrontato 30 (6x5) caratteri")
15
AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW AAKKQW AAKQW 1 2 3 4 5 6 7 8 9 10 Numero degli allineamenti generati 1 3 6 10 15 20 24 27 29 30 Numero dei confronti tra residui effettuati

16
calcolo della similarità tra due sequenze date un programma per calcolatore ha un tempo di esecuzione che in generale dipende dal numero di operazioni necessarie per eseguirlo un programma che calcoli la similarità di sequenza tra due sequenze date deve effettuare un numero di confronti che dipende dal prodotto della lunghezza delle due sequenze O(nm) ~ O(n 2 )
 ~ O(n 2 )")
17
Allineamento semplice Lallineamento semplice si ottiene facendo scorrere una sequenza sullaltra un nucleotide alla volta (passo 1) CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | | | | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | |||| | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | ||| | || || CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | | || || CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || || || | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC ||||||||||||||||||||||||||||| CGAAATCGCATCAGCATACGATCGCATGC
 CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | | | | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | |||| | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | ||| | || || CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | | || || CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || || || | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | | CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || CGAAATCGCATCAGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC ||||||||||||||||||||||||||||| CGAAATCGCATCAGCATACGATCGCATGC")
18
Quando si deve effettuare una ricerca per similarità di sequenza in una banca dati, loperazione di confronto tra due sequenze deve inoltre essere ripetuta per ogni coppia di sequenze: 1) sequenza in input (query sequence) 2) ognuna delle sequenze della banca dati ricerca per similarità in una banca dati
 sequenza in input (query sequence) 2) ognuna delle sequenze della banca dati ricerca per similarità in una banca dati")
19
esiste quindi una forte necessità di utilizzare algoritmi RAPIDI!

20
ricerca per similarità in una banca dati … e non abbiamo ancora considerato tutte le ulteriori complicazioni legate alla possibile presenza di gaps nellallineamento tra due sequenze!

21
calcolo della similarità tra due sequenze date necessità di trattare linserimento e/o la delezione di caratteri (gaps) IPLMTRWDQEQESDFGHKLPIYTREWCTRG |||||||||| CHKIPLMTRWDQQESDFGHKLPVIYTREW 10 25 IPLMTRWDQEQESDFGHKLP-IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVIYTREW
 IPLMTRWDQEQESDFGHKLPIYTREWCTRG |||||||||| CHKIPLMTRWDQQESDFGHKLPVIYTREW IPLMTRWDQEQESDFGHKLP-IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVIYTREW")
22
calcolo della similarità tra due sequenze date linserimento di gaps comporta una modifica del nostro iniziale SEMPLICE metodo di misura della similarità possiamo associare un punteggio di penalizzazione (gap penalty) per ogni gap aggiunto allallineamento o attribuire un punteggio di penalizzazione diverso per lapertura di un gap nellallineamento o per il suo allungamento (gap extension penalty)
 per ogni gap aggiunto allallineamento o attribuire un punteggio di penalizzazione diverso per lapertura di un gap nellallineamento o per il suo allungamento (gap extension penalty)")
23
calcolo della similarità tra due sequenze date gap extension penalty (es.: -0.1 per ogni ins/del successiva alla prima) IPLMTRWDQEQESDFGHKLP----IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVGSSIYTREW gap creation penalty (es.: -1 per ogni gap) IPLMTRWDQEQESDFGHKLP-IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVIYTREW
 IPLMTRWDQEQESDFGHKLP----IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVGSSIYTREW gap creation penalty (es.: -1 per ogni gap) IPLMTRWDQEQESDFGHKLP-IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVIYTREW")
24
Allineamento con gaps Lallineamento semplice non sempre funziona bene CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | | | | | | | | CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | ||||| | | | CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | | || || CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | ||| | | || || CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || | || | CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || || | | CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC | | | CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC || |||||||||||||||| CGAAATCGCATCACGCATACGATCGCATGC CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC ||||||||||||| | CGAAATCGCATCACGCATACGATCGCATGC A meno che le sequenze non coincidano perfettamente è molto spesso necessario introdurre gaps CGCTTCGGACGAAATCGCATCA-GCATACGATCGCATGCCGGGCGGGATAA ||||||||||||| |||||||||||||||| CGAAATCGCATCACGCATACGATCGCATGC

25
calcolo della similarità tra due sequenze date un algoritmo di allineamento che tenesse conto 1)del possibile inserimento di un gap in ogni possibile posizione delle due sequenze e 2)di ogni possibile lunghezza di un gap in ogni possibile posizione sarebbe estremamente LENTO
del possibile inserimento di un gap in ogni possibile posizione delle due sequenze e 2)di ogni possibile lunghezza di un gap in ogni possibile posizione sarebbe estremamente LENTO")
26
calcolo della similarità tra due sequenze date da ciò discende la necessità di trovare soluzioni alternative, ovvero nuovi algoritmi per lallineamento delle sequenze

27
Matrici di punti - dot matrices consideriamo una matrice in cui la prima riga in alto coincide con la sequenza1 scritta da sinistra a destra e in cui la prima colonna a sinistra coincide con la sequenza2 scritta dallalto in basso in ogni punto in cui la sequenza nella riga e nella colonna coincidono, introduciamo un asterisco nel grafico. Le regioni delle sequenze che possono essere allineate senza introdurre gap emergono come una serie contigua di punti sulla diagonale

28
calcolo della similarità tra due sequenze date - Uso di matrici di punti (DOT MATRICES) m a r g a r e t d y h o f f a margaretdayhoffoelkya * *** ** * * **** *** ** ** * * * * * * * *
 m a r g a r e t d y h o f f a margaretdayhoffoelkya * *** ** * * **** *** ** ** * * * * * * * *")
29
le dot matrices esprimono una buona rappresentazione grafica di un allineamento consentono di visualizzare similarità di sequenza anche in presenza di gaps, che appaiono come salti di diagonale esistono programmi in grado di sfruttare gli schemi tipo dot matrix per valutare la similarità tra sequenze e identificare il miglior allineamento

30
il rumore di fondo é molto alto perché molti dei match tra sequenze costruiti in questo modo sono casuali e dipendono da singole occorrenze dello stesso residuo in posizioni diverse delle due sequenze. possiamo calcolare il numero di match in una finestra, per esempio di 5 o 15 residui / basi, e decidere di introdurre un punto nel grafico solo se una certa percentuale minima di questi (es. 50%) sono identici. il punto viene posizionato al centro della finestra. Matrici di punti - dot matrices
 sono identici. il punto viene posizionato al centro della finestra. Matrici di punti - dot matrices.")
31
calcolo della similarità tra due sequenze date - Uso di matrici di punti (DOT MATRICES)
")
32
Le ricerche di similarità si basano sul confronto sistematico di una sequenza di partenza (generalmente chiamata query) con ognuna delle sequenze del database. Considerando che molti database di sequenze, come ad esempio GenBank contengono oltre un milione di sequenze diverse è necessario disporre di programmi in grado di portare avanti queste ricerche in modo automatico. Alcuni programmi, come BLAST, sono estremamente efficienti e sono in grado di portare a termine una ricerca di similarità in pochi secondi. Questi programmi si basano essenzialmente su procedure che: * per ogni sequenza del database * effettua l'allineamento; * attribuisce un punteggio; * se il punteggio è alto lo memorizza; * ripete fino alla fine delle sequenze; * mostra la lista dei migliori allineamenti trovati

33
E' importante notare che è essenziale attribuire un punteggio agli allineamenti, altrimenti non si avrebbe modo di stabilire se un allineamento è migliore di un altro. Deve essere notato che con l'approccio della dot matrix non viene trovato alcun allineamento, ma viene semplicemente mostrata una matrice che deve essere interpretata visivamente. Quindi per effettuare ricerche di similarità questo semplice approccio non e sufficiente: abbiamo bisogno di un sistema che sia anche in grado di trovare automaticamente gli allineamenti migliori. Molto schematicamente il problema degli allineamenti viene risolto da programmi che sono in grado di identificare il "percorso" migliore all'interno di una dot matrix. Cioè il percorso che totalizza il massimo punteggio. Per percorso si intende l'insieme di caselle che corrispondono agli aminoacidi appaiati.

34
calcolo della similarità tra due sequenze date - le matrici di sostituzione nel caso delle sequenze proteiche, gli allineamenti e il calcolo della similarità possono essere notevolmente migliorati dallintroduzione di diversi schemi di punteggio, noti come MATRICI DI SOSTITUZIONE che comprendono punteggi diversi da 0 e da 1 per lappaiamento di residui amminoacidici

35
Molto semplicemente, il punteggio può essere calcolato attribuendo un valore positivo ad ogni lettera appaiata correttamente e una penalità ad ogni gap. La somma di questi valori rappresenta quindi il punteggio dell'intero allineamento. Nel caso più semplice possiamo assegnare il valore di '1' ad ogni identità e '0' ad ogni "mismatch" cioè ai caratteri non appaiati correttamente. Un tale criterio non è però molto valido perchè non ha senso penalizzare tutti i mismatch allo stesso modo. Ad esempio la leucina e l'isoleucina sono molto simili; quindi se questi due aminoacidi vengono appaiati possiamo pensare che viene rispettato un certo criterio di similarità, anche se non c'è identità assoluta. Estendendo questo ragionamento possiamo attribuire ad ogni possibile coppia di aminoacidi un punteggo di appaiamento. In questo modo otteniamo una matrice di sostituzione.

36
le matrici di sostituzione potremmo raggruppare residui in classi a seconda della similarità delle loro caratteristiche chimico- fisiche, e sommare 1 al punteggio per appaiamenti di residui della stessa classe e sottrarre 1 per residui di classi diverse I L V G A E D F Y W R K H C P

37
le matrici di sostituzione Margaret Dayhoff raccolse statistiche sulle frequenze di sostituzioni amminoacidiche nelle sequenze proteiche allora note via via che le sequenze divergono, le mutazioni si accumulano per misurare la probabilità relativa di una particolare sostituzione (per esempio Asp--> Glu) possiamo contare quanti Asp sono diventati Glu in allineamenti di sequenze omologhe
 possiamo contare quanti Asp sono diventati Glu in allineamenti di sequenze omologhe")
38
le matrici di sostituzione è comunque necessario evitare di considerare allineamenti in cui possano essere avvenuti sostituzioni multiple in determinate posizioni per cui questi calcoli devono venire effettuati su coppie di sequenze MOLTO SIMILI tra loro, in modo da poter assumere che nessuna posizione è mutata più di una volta la divergenza di due sequenze si può misurare in PAM: 1 PAM = 1 Percent Accepted Mutation

39
le matrici di sostituzione due sequenze sono separate da 1 PAM se hanno il 99% di identità la frequenza delle sostituzioni amminoacidiche può essere calcolata in coppie di sequenze poco divergenti (1 PAM) frequenze di sostituzioni amminoacidiche per sequenze più divergenti possono essere calcolate a partire da queste, fino ad ottenere matrici PAM 250, corrispondenti a similarità di sequenza del 20%
 frequenze di sostituzioni amminoacidiche per sequenze più divergenti possono essere calcolate a partire da queste, fino ad ottenere matrici PAM 250, corrispondenti a similarità di sequenza del 20%")
40
PAM 0 30 80 110 200 250 % identità 100 75 60 50 25 20 le matrici di sostituzione se due sequenze sono filogeneticamente distanti è opportuno usare matrici PAM con indici più alti, e viceversa

41
Il tutto si può facilmente rappresentare con una matrice di 20x20, quattrocento valori. La seguente tabella corrisponde alla matrice PAM250

42
A R N D C Q E G H I L K M F P S T W Y V B Z A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 BLOSUM 62

43
Allineamento globale: LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||. | | |.|.| || || | || TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG Allineamento locale: TGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||||||||.||||| TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHK Allineamento globale o locale?

44
Allineamento globale: LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||. | | |.|.| || || | || TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG Allineamento locale: TGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||||||||.||||| TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHK Allineamento globale o locale? id. 14 13 sost. cons. 3 1 Ma qual è lallineamento MIGLIORE?

45
Allineamento globale o locale? 1) scegliamo il miglior allineamento dal punto di vista biologico, e poi… 2) cerchiamo il modo di privilegiarlo dal punto di vista computazionale spesso gli allineamenti locali hanno una migliore rispondenza con la realtà funzionale
 scegliamo il miglior allineamento dal punto di vista biologico, e poi… 2) cerchiamo il modo di privilegiarlo dal punto di vista computazionale spesso gli allineamenti locali hanno una migliore rispondenza con la realtà funzionale.")
46
Allineamento globale o locale? similarità locali servono a identificare proteine anche diverse, ma che contengono lo stesso dominio

47
Allineamento globale o locale? a livello di DNA, troviamo regioni con similarità locali che riflettono situazioni interessanti: ad esempio introni/esoni, inserzioni/delezioni, transposoni, regioni promotore… esoniintroni

48
gli allineamenti globali possono comunque essere utilizzati per confrontare accuratamente due sequenze la cui similarità sia estesa per tutta la lunghezza. Allineamento globale o locale?

49
allineamenti tra due sequenze abbiamo definito un nuovo schema di punteggi per la valutazione della similarità tra due sequenze una matrice di sostituzione per valutare lappaiamento tra qualsiasi coppia di residui penalizzazioni appropriate per lapertura o lestensione di un gap

50
allineamenti tra due sequenze ci serve ora un metodo (un algoritmo) per generare il miglior allineamento possibile tra due sequenze, tenendo anche conto delle possibili inserzioni e delezioni un algoritmo famoso per lallineamento tra due sequenze si basa su una tecnica matematica nota come programmazione dinamica
 per generare il miglior allineamento possibile tra due sequenze, tenendo anche conto delle possibili inserzioni e delezioni un algoritmo famoso per lallineamento tra due sequenze si basa su una tecnica matematica nota come programmazione dinamica")
51
allineamenti tra due sequenze - programmazione dinamica buone notizie: il metodo trova il miglior allineamento globale tra due sequenze cattive notizie: 1) spesso loutput mostra più allineamenti DIVERSI col massimo del punteggio 2) ci mette TROPPO TEMPO per effettuare una ricerca esaustiva
 spesso loutput mostra più allineamenti DIVERSI col massimo del punteggio 2) ci mette TROPPO TEMPO per effettuare una ricerca esaustiva")
52
Algoritmi dinamici di allineamento per effettuare un allineamento è prima di tutto necessario scegliere una matrice di sostituzione per valutare gli appaiamenti tra residui e definire dei punteggi di penalizzazione per i gaps algoritmi di allineamento che utilizzano una tecnica di programmazione dinamica: Needleman e Wunsch (1970) Smith e Waterman (1981)
 Smith e Waterman (1981)")
53
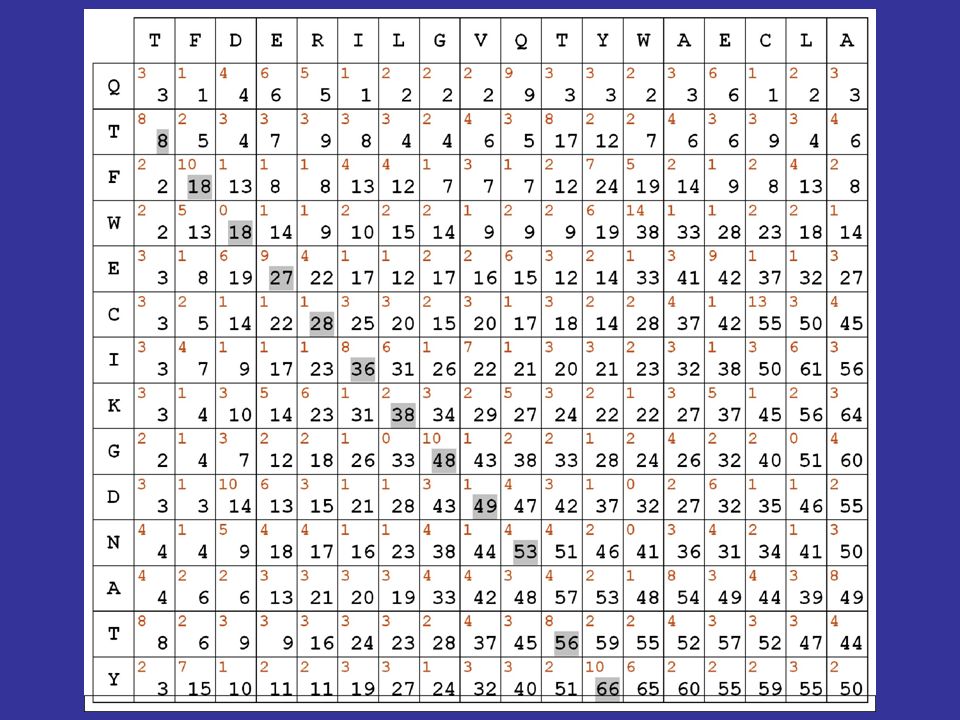
Algoritmi dinamici di allineamento dividiamo la procedura in 3 passi successivi 1) consideriamo le due sequenze da allineare in una specie di dot matrix : nelle caselle scriviamo i punteggi in rosso derivati dalla matrice di sostituzione scelta
 consideriamo le due sequenze da allineare in una specie di dot matrix : nelle caselle scriviamo i punteggi in rosso derivati dalla matrice di sostituzione scelta")
55
calcolando le somme lungo le diagonali, effettueremmo unoperazione equivalente al calcolo dei punteggi ottenuto facendo scorrere le due sequenze luna sullaltra resterebbe aperto il problema della valutazione dei gaps Algoritmi dinamici di allineamento

56
passo 2 della procedura: ricerca del percorso che consente di ottenere il massimo punteggio in base a delle regole stabilite, tenendo anche conto dei gaps Algoritmi dinamici di allineamento qualche semplice osservazione ci consente di risparmiare tempo di calcolo:

57
se una sequenza è scritta da sinistra a destra e laltra dall'alto in basso, allora qualsiasi percorso valido deve mantenere sempre una direzione tendenziale che va dall'angolo in alto a sinistra a quello in basso a destra Algoritmi dinamici di allineamento

58
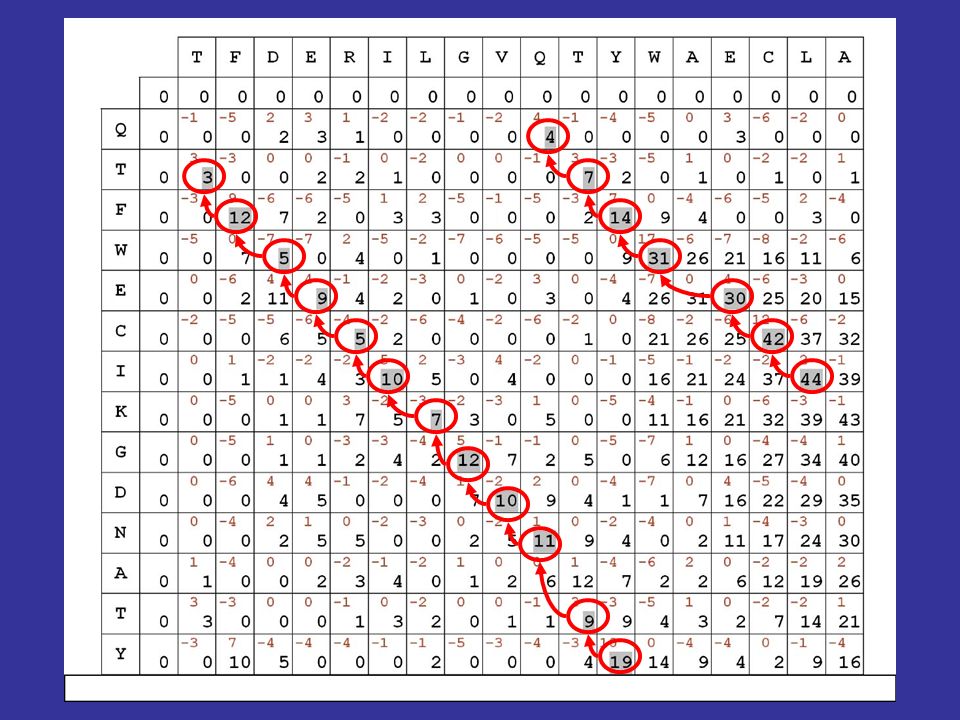
Misura della similarità: i punteggi in diagonale si SOMMANO fuori dalla diagonale, si PENALIZZA di 5 punti

59
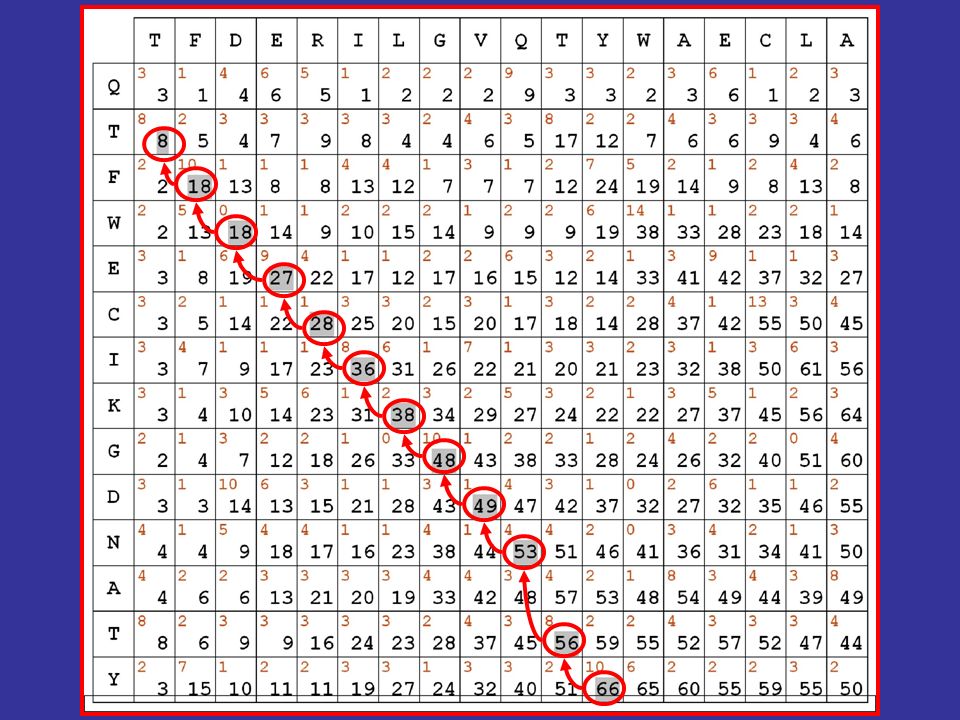
vediamo quindi come si calcolano le cifre riportate in nero nella matrice evidenzieremo poi in grigio il migliore percorso allinterno della matrice, secondo le regole e i punteggi stabiliti Algoritmi dinamici di allineamento

61
il migliore allineamento globale per le sequenze in matrice risulta quindi il seguente: TFDERILGVQ-TYWAECLA || | | |. || QTFWECIKGDNATY Algoritmi dinamici di allineamento

62
il fatto di aver usato matrici di sostituzione contenenti esclusivamente valori positivi fa sì che il valore massimo della matrice si trovi sempre nellultima riga o nellultima colonna ne consegue che lallineamento ottenuto è un allineamento globale

63
Algoritmi dinamici di allineamento la procedura descritta corrisponde quasi esattamente allalgoritmo per lallineamento globale pubblicato da Needleman e Wunsch nel 1970

64
se le matrici contenessero invece sia valori positivi che negativi (come le pam viste nel corso della scorsa lezione), i valori più alti potrebbero trovarsi anche in porzioni INTERNE alla matrice e descrivere di conseguenza allineamenti locali Algoritmi dinamici di allineamento
, i valori più alti potrebbero trovarsi anche in porzioni INTERNE alla matrice e descrivere di conseguenza allineamenti locali Algoritmi dinamici di allineamento")
66
TFDERILGVQ-TYWAECLA || | | |. || QTFWECIKGDNATY Allineamento globale TFDERILGVQTYWAECLA ||.| ||. QTFW-ECIKGDNATY Allineamento locale

67
Algoritmi dinamici di allineamento é importante notare che il punteggio massimo lo è in senso relativo il valore assoluto dei punteggi che associamo agli allineamenti dipende dai valori contenuti nella matrice di sostituzione che utilizziamo ci sono matrici i cui valori variano tra 0 e 100, altre che variano da -1 a +1, adottando punteggi decimali

68
Algoritmi dinamici di allineamento L algoritmo di Needleman e Wunsch è stato sviluppato per lallineamento globale L algoritmo di Smith e Waterman è stato sviluppato per lallineamento locale Ma ciò che realmente fa diventare un algoritmo di questo tipo locale o globale è il tipo di matrice di sostituzione che si usa: se contiene solo valori + allineamenti globali se contiene valori +/- allineamenti locali

69
Metodi euristici per lallineamento gli algoritmi descritti effettuano delle ricerche esaustive ed esplorano tutto lo spazio degli allineamenti possibili si tratta comunque di algoritmi di ordine n 2, ovvero per allineare due sequenze lunghe ognuna 1000 residui, effettuano 1000x1000 = un milione di confronti

70
Metodi euristici per lallineamento per effettuare ricerche di similarità in banche dati, cè comunque necessità di algoritmi più veloci PGM allineamento similarità di sequenza seq1 seq2 PGM lista di proteine simili alla query seq DB

71
la crescita esponenziale delle dimensioni delle banche dati di sequenze biologiche ha portato allo sviluppo di programmi (come FASTA e BLAST) in grado di effettuare velocemente ricerche di similarità, grazie a soluzioni euristiche che, come vedremo, sono basate su assunzioni non certe, ma estremamente probabili. Metodi euristici per lallineamento in pratica la ricerca è resa più veloce a scapito della certezza di avere veramente trovato la soluzione migliore.

72
calcolo di punteggi con matrice di sostituzione, selezione dei migliori punteggi Metodi euristici per lallineamento (FASTA) ricerca di parole contigue per sola identità sulla stessa diagonale
 ricerca di parole contigue per sola identità sulla stessa diagonale")
73
applicazione di S&W su una stretta banda per ottimizzare lallineamento Metodi euristici per lallineamento (FASTA) unione di frammenti che possono essere congiunti entro una soglia di accettabilità
 unione di frammenti che possono essere congiunti entro una soglia di accettabilità")
74
Metodi euristici per lallineamento (FASTA) FASTA è stato pubblicato nel 1985 da Lipman e Pearson
 FASTA è stato pubblicato nel 1985 da Lipman e Pearson")
75
Allineamento multiplo di sequenze PGM allineamento similarità di sequenza seq1 seq2 PGM lista di proteine simili alla query seq DB é possibile utilizzare una lista di sequenze simili tra loro per generare un ALLINEAMENTO MULTIPLO DI SEQUENZE













>")
>")




 Algoritmi di allineamento 2) Algoritmi di ricerca in database>")

