Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Base molecolare della variabilità
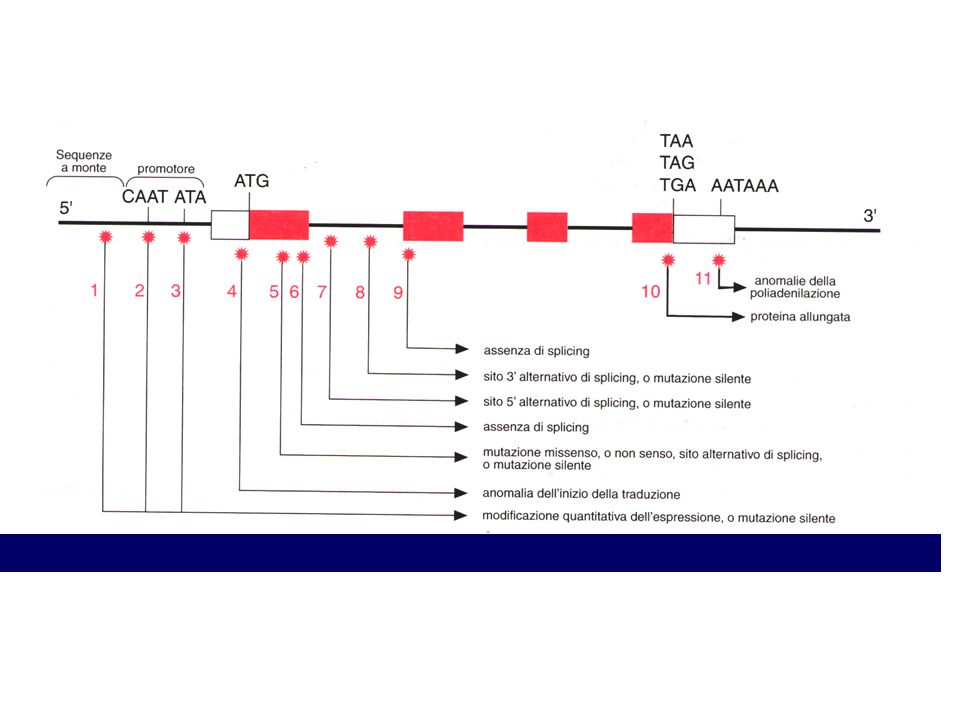
MUTAZIONE Cambiamento della struttura del genoma che causa una variazione del genotipo attraverso le generazioni Base molecolare della variabilità

2
Comparsa improvvisa di un caso di acondroplasia da genitori sani
Condizione autosomica dominante Gene coinvolto "recettore del fattore di crescita dei fibroblasti di tipo 3", (FGFR3). Questo recettore, in condizioni normali esplica un controllo di tipo negativo sulle cellule della cartilagine di accrescimento. Mutazioni di FGFR3 lo rendono costitutivamente attivato (mutazioni con “guadagno di funzione”) FGFR3 mutato è in grado di segnalare continuamente alla cellula un segnale negativo, col risultato di inibire l’allungamento dell’osso.
. Questo recettore, in condizioni normali esplica un controllo di tipo negativo sulle cellule della cartilagine di accrescimento. Mutazioni di FGFR3 lo rendono costitutivamente attivato (mutazioni con guadagno di funzione ) FGFR3 mutato è in grado di segnalare continuamente alla cellula un segnale negativo, col risultato di inibire l’allungamento dell’osso.")
3
FGFR3 CROUZON TD ACONDROPLASIA TD IPOCONDROPLASIA S - S Ala(391)Glu
1138 G A 1138 G C Gly (380)Arg D. transmembranoso CROUZON Ala(391)Glu D. transmembranario IPOCONDROPLASIA Lys(540)Leu 70% TK1 TD Arg(258)Cys IgII-IgIII interloop TD Lys(650)Met TK2
Arg. D. transmembranoso. CROUZON. Ala(391)Glu. D. transmembranario. IPOCONDROPLASIA. Lys(540)Leu 70% TK1. TD. Arg(258)Cys IgII-IgIII. interloop. TD. Lys(650)Met TK2.")
4
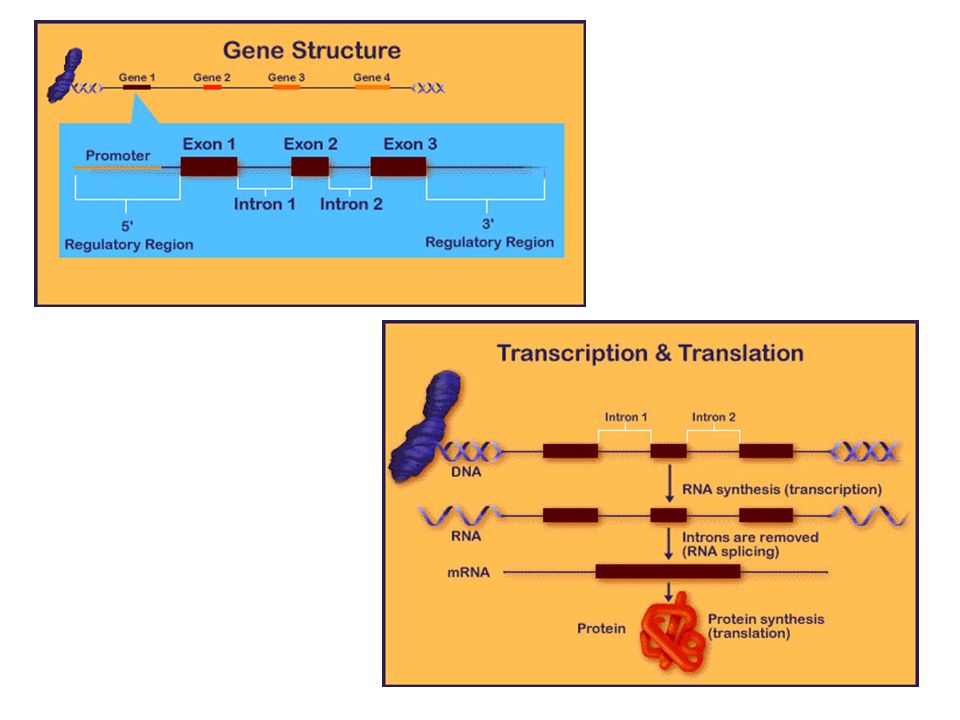
Mutazione in un gene Evento mutazionale
Gene normale Prodotto genico normale Espressione fenotipica normale Trascrizione e traduzione Evento mutazionale Prodotto genico parzialmente funzionante/non funzionante/assente Espressione fenotipica alterata Gene mutato N.B. NON TUTTE LE MUTAZIONI CAUSANO UN’ALTERAZIONE DELLE PROTEINE E NON TUTTE LE MUTAZIONI AVVENGONO NELLA REGIONE CODIFICANTE DEL GENE

5
mutazioni …..EFFETTO MINIMO O NULLO POLIMORFISMI LETALE
(non alterazioni della proteina) POLIMORFISMI se la frequenza è superiore all’1% LETALE proteina anomala FENOTIPO ANOMALO
 POLIMORFISMI. se la frequenza è superiore all’1% LETALE. proteina anomala FENOTIPO ANOMALO.")
6
Classificazione in base ……….. …….. alla tipologia e all’estensione
Mutazioni cromosomiche: sono alterazioni di numero o di struttura dei cromosomi Mutazioni geniche: interessano i geni TIPO MECCANISMO FREQUENZA ESEMPIO Genomiche Errori di segregazione /div. cell Aneuploidia N° cromosomi Cromosomiche Errori di ricombinazione 6X10-4/div. cell Duplicazioni Struttura cromosomi Delezioni Geniche Errori della duplicazione /bp/div. cell Mut. puntiformi Poche basi / locus/gen.

7
Mutazioni spontanee: Duplicazione. Ricombinazione. Segregazione.
3 miliardi di coppie di basi devono essere replicate ad ogni divisione cellulare, in un periodo di 2-3 ore. Velocità di circa bp/sec. Avvengono casualmente degli errori Ricombinazione. Processo fondamentale per generare “diversità biologica”, a causa della struttura di particolari regioni genomiche, è un’altra fonte di possibili errori. Segregazione. Processo finale della mitosi e della meiosi. Avvengono casualmente degli errori che portano ad una errata ripartizione dei cromosomi nelle cellule figlie.

8
COSA CAUSA LE MUTAZIONI?
Classificazione in base …… …….. all’origine Mutazioni spontanee: insorgono in assenza di agenti mutageni esterni e sono prodotte da fenomeni di ricombinazione (crossing-over) o replicazione del DNA Mutazioni indotte: dovute all’azione sul DNA di agenti chimici, fisici o biologici
 o replicazione del DNA. Mutazioni indotte: dovute all’azione sul DNA di agenti chimici, fisici o biologici.")
9
La differenza principale tra mutazioni spontanee ed indotte è la frequenza di mutazione
Numero di eventi mutazionali mutazionali (cellule o individui) Totale della popolazione Le mutazioni indotte hanno una frequenza di mutazione superiore a quella che si osserva per le mutazioni spontanee
 Totale della popolazione. Le mutazioni indotte hanno una frequenza di mutazione superiore a quella che si osserva per le mutazioni spontanee.")
11
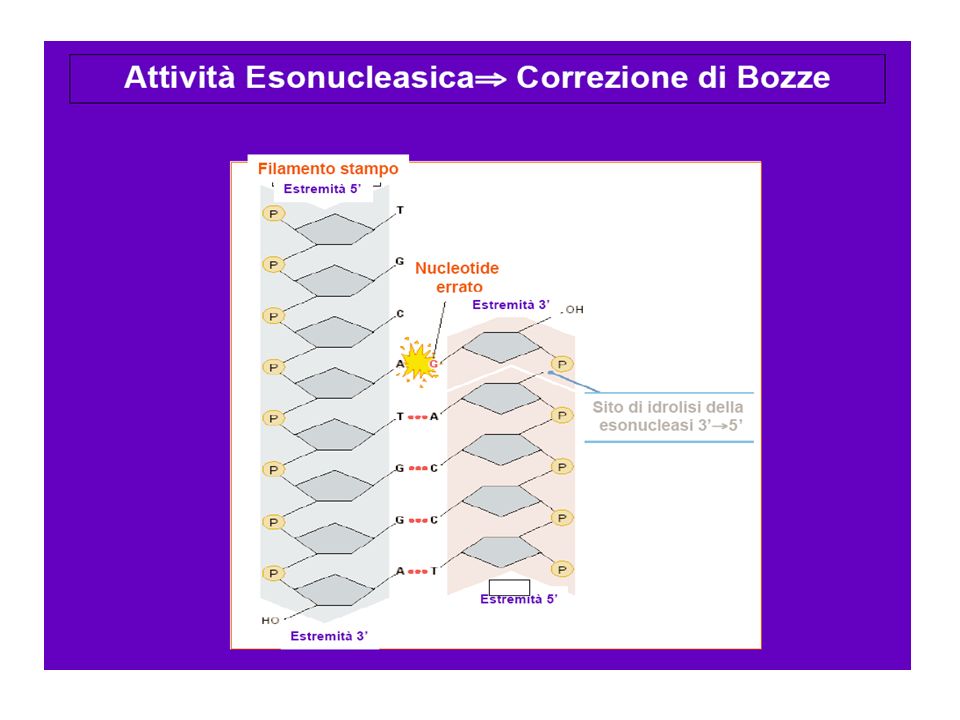
MUTAZIONI PUNTIFORMI Molti fattori fisici e chimici, endogeni ed esogeni, tendono se permanenti ad alterare il DNA genomico. Spesso le lesioni prodotte da: rottura di una catena creazione di legami covalenti tra basi contigue o complementari escissione di una o più basi errore di replicazione sono riconosciute e riparate dai sistemi enzimatici deputati a riparare il DNA. Se ciò non avviene l’alterazione che si verifica su una catena può essere fissata al momento della replicazione: si tratta allora di una mutazione puntiforme che si tramanderà nelle generazioni cellulari successive.

12
Mutazioni indotte: Calore Radiazioni UV
fattori esogeni (mutageni chimici, ionizzanti e irradiazione UV, calore) Calore Radiazioni UV Il calore provoca idrolisi dei legami b-N-glicosidici, creando un sito senza base. Lo zucchero-fosfato rimanenti vengono facilmente persi lasciando così un “buco”. Il risultato è generalmente una delezione.
 Calore. Radiazioni UV. Il calore provoca idrolisi dei legami b-N-glicosidici, creando un sito senza base. Lo zucchero-fosfato rimanenti vengono facilmente persi lasciando così un buco . Il risultato è generalmente una delezione.")
13
(HOT SPOTS MUTAZIONALI)
Le mutazioni non sono distribuite casualmente ed uniformemente nel genoma. L’incidenza delle mutazioni nei diversi loci dipende da diversi fattori: - dimensione del locus considerato - capacità codificante o meno - caratteristiche di sequenza. ZONE CALDE DI VARIABILITA’ (HOT SPOTS MUTAZIONALI)
")
14
Grandezza dei geni

15
Frequenza di mutazione
di alcuni geni Gene Nuove mutazioni per 106 gameti Emofilia B – 3 Aniridia – 5 Retinoblastoma – 12 Acondroplasia – 40 Emofilia A – 57 Distrofia muscolare di Duchenne 43 – 105 Neurofibromatosi – 100 Polycystic kidney disease – 120 La frequenza di mutazione (mutazione / gene / divisione cellulare) può variare da 10-4 a a seconda della grandezza del gene e del fatto che vi siano “hot spots” mutazionali (la frequenza in molti loci è compresatra 10-5 e 10-6).
 può variare da 10-4 a 10-7 a seconda della grandezza del gene e del fatto che vi siano hot spots mutazionali (la frequenza in molti loci è compresatra 10-5 e 10-6).")
19
MUTAZIONI PUNTIFORMI Cause più frequenti di malattie genetiche (circa il 70%). Possono essere causate da: alterazioni biochimiche e soprattutto da deaminazione di citosine metilate localizzate in siti particolari che contengono doppiette CG (punti caldi di mutazione: HOT SPOTS) errori di replicazione o di riparazione del DNA (delezione o addizione di un numero molto piccolo di basi). Incidenti di replicazione si producono soprattutto a livello di sequenze corte ripetute.
 errori di replicazione o di riparazione del DNA (delezione o addizione di un numero molto piccolo di basi). Incidenti di replicazione si producono soprattutto a livello di sequenze corte ripetute.")
20
TRASFORMAZIONE DELLA CITOSINA IN TIMINA
NH2 3HC N NH2 O O 3HC N NH metilazione deaminazione O O N N TIMINA CITOSINA 5’-METILCITOSINA

21
BASI MOLECOLARI DELLE MUTAZIONI SPONTANEE ………… APPAIAMENTO ERRATO
Errori che intervengono nei meccanismi di replicazione e di riparazione del DNA

22
BASI MOLECOLARI DELLE MUTAZIONI SPONTANEE
Il fenomeno dello slittamento della forca replicativa è spesso la causa di brevi inserzioni/delezioni Replicazione normale Corte sequenze ripetute in tandem Slittamento indietro In questo caso si possono verificare inserzioni/delezioni per appaiamento errato causato da scivolamento di un filamento Slittamento avanti

23
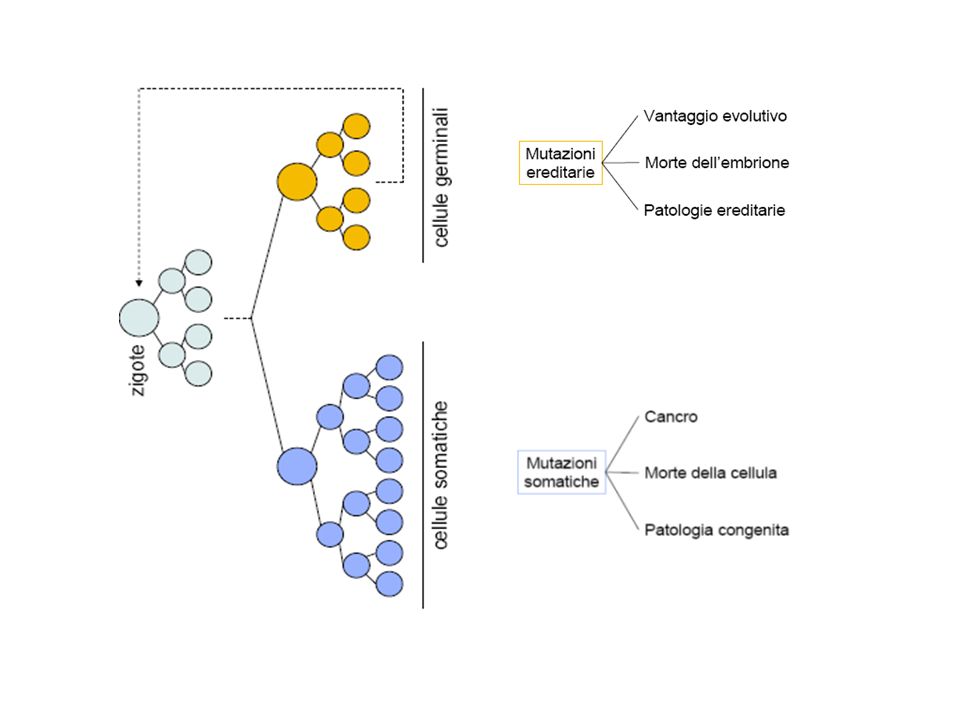
Classificazione in base ……… …….. alla sede
Mutazioni germinali Si verificano nei gameti e possono essere trasmesse alla prole.Danno origine ad un individuo mutato sia nelle cellule somatiche sia nei gameti Mutazioni somatiche Sono presenti esclusivamente nelle cellule dell’organismo (cellule somatiche) e non nei gameti, pertanto non possono essere trasmesse alla discendenza. Le caratteristiche mutate si manifestano solo nell’individuo in cui e’ avvenuta la mutazione e non vengono trasmesse alle generazioni successive.
 e non nei gameti, pertanto non possono essere trasmesse alla discendenza. Le caratteristiche mutate si manifestano solo nell’individuo in cui e’ avvenuta la mutazione e non vengono trasmesse alle generazioni successive.")
25
TIPO DI MUTAZIONE (può interessare uno o pochi nucleotidi oppure centinaia di milioni di nucleotidi): Delezioni Inserzioni Duplicazioni Inversioni Sostituzioni Transizioni purina <--> purina pirimidina <--> pirimidina Trasversioni purina<-->pirimidina
: Delezioni Inserzioni Duplicazioni Inversioni Sostituzioni Transizioni purina <--> purina pirimidina <--> pirimidina Trasversioni purina<-->pirimidina")
26
TRANSIZIONI Una purina (A o G) e’ sostituita con un’altra purina (G o A) o una pirimidina (C o T) con l’altra pirimidina (T o C) ATTTCGCCGAATTGC TRANSIZIONE ATTTCGCCGGATTGC TAAAGCGGCTTAACG TAAAGCGGCCTAACG

27
TRANSVERSIONI Una purina (A o G) e’ sostituita con una pirimidina (C o T) e viceversa ATTTCGCCGAATTGC TRANSVERSIONE ATTTCGCCGCATTGC TAAAGCGGCTTAACG TAAAGCGGCGTAACG

28
MUTAZIONI ….. EFFETTO Silente nessun cambiamento nell’aminoacido codificato Missenso conservativa = residuo aa stessa classe non-conservativa = residuo aa classe diversa Nonsense terminazione prematura della proteina Frameshift cambiamenti nel frame di lettura della sequenza

29
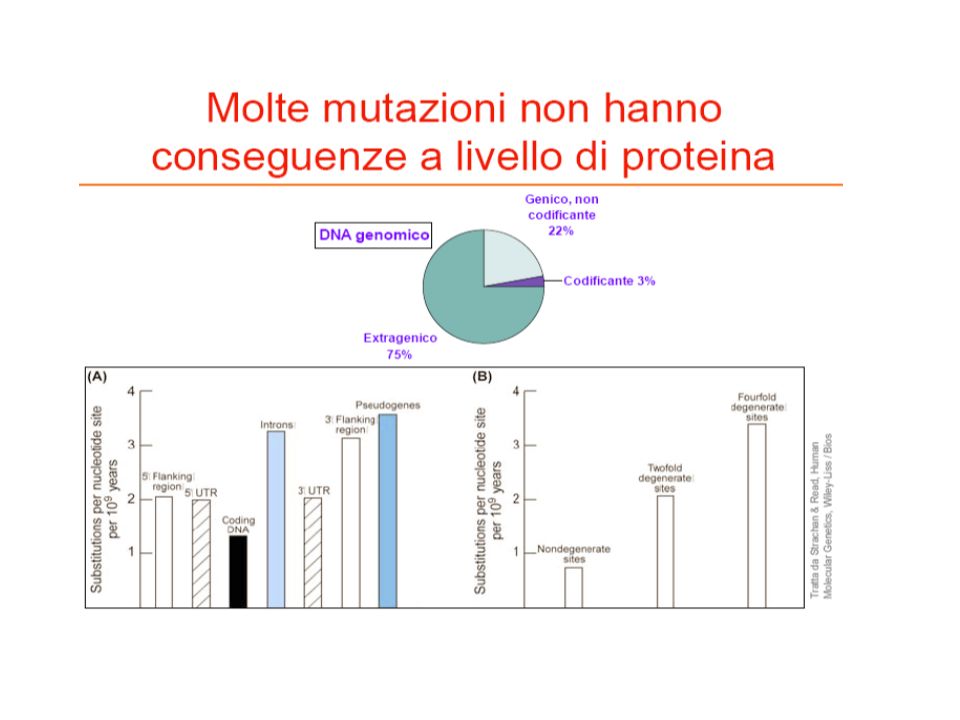
Molte mutazioni non hanno conseguenze a livello di proteina

30
Molte mutazioni non hanno conseguenze a livello di proteina

31
AMINOACIDI Amminoacidi neutri con catene non polari
Amminoacidi neutri con catene polari Amminoacidi acidi con catene polari Amminoacidi basici con catene polari

32
Amminoacido neutro con catene polari Amminoacido neutro con catene polari Amminoacido neutro con catene non polari

34
CODICE GENETICO

35
MUTAZIONI SILENTI

36
MUTAZIONI NELLE REGIONI CODIFICANTI MUTAZIONI NONSENSO
Risultano da una sostituzione nucleotidica che comporta la formazione di un codone di stop (UAA, UAG o UGA). EFFETTI ….. determinano l’interruzione della traduzione, e la proteina finale, che è troncata, non è generalmente espressa.
. EFFETTI ….. determinano l’interruzione della traduzione, e la proteina finale, che è troncata, non è generalmente espressa.")
37
MUTAZIONI NELLE REGIONICODIFICANTI MUTAZIONI MISSENSO modificano il significato di un codone.
Risulta un cambiamento di un aminoacido nella proteina corrispondente. L’impatto finale è variabile a seconda della natura del cambiamento e della sua localizzazione sulla catena polipeptidica. La mutazione può avere effetto sulla funzione e/o sulla stabilità della proteina; al contrario può non avere alcuna influenza.

38
D167H (ASPARTATO 167 ISTIDINA)
aa neutro con catene non polari aa neutro con catene polari

39
MUTAZIONI NELLE REGIONI CODIFICANTI MUTAZIONI MISSENSO modificano il significato di un codone.
Se la mutazione si verifica a carico di uno dei 3 codoni di stop (UAA, UAG, UGA) può determinare un proseguimento della traduzione fino al codone di stop successivo. Ne risulta un allungamento della catena polipeptidica.
 può determinare un proseguimento della traduzione fino al codone di stop successivo. Ne risulta un allungamento della catena polipeptidica.")
40
MUTAZIONI CHE CAMBIANO LA FASE DI LETTURA MUTAZIONI FRAMESHIFT
Qualunque inserzione o delezione (in un esone) di un numero di basi che non sia multiplo di 3 comporta la modifica della fase di lettura. Determina la comparsa di un codone di stop prematuro.
 di un numero di basi che non sia multiplo di 3 comporta la modifica della fase di lettura. Determina la comparsa di un codone di stop prematuro.")
41
Cambiamento della fase di lettura del codice
FRAMESHIFT Delezione di un aa Cambiamento della fase di lettura del codice

42
MUTAZIONI CHE ALTERANO LO SPLICING
sito donatore di splicing sito accettore di splicing mRNA GT AG GT AG SPLICING MUTAZIONI CHE ALTERANO LO SPLICING La mutazione colpisce una sequenza consenso dello splicing …….. il processo di eliminazione degli introni non può avvenire correttamente

43
sito donatore di splicing
sito accettore di splicing mRNA GT AG GT AG SPLICING Mutazioni nei siti canonici GTe AG localizzati all’inizio e alla fine dell’introne. Mutazioni nelle sequenze consenso importanti per il riconoscimentodei limiti dell’introne da parte dei fattori di splicing Sostituzione di una base in una sequenza intronica o esonica che presenti un certo grado di omologia con sequenze consenso dello splicing (SITI CRIPTICI DI SPLICING)
")
44
MUTAZIONI CHE MODIFICANO SEGNALI IMPORTANTI PER I SITI DI SPLICING
OMISSIONE DI UN ESONE GT AG CG SD SA

45
MUTAZIONI CHE MODIFICANO SEGNALI IMPORTANTI PER I SITI DI SPLICING
RITENZIONE DI UN INTRONE SD SA SD SA GT CG GT AG GT CT AG SD SA

46
MUTAZIONI CHE MODIFICANO SEGNALI IMPORTANTI PER I SITI DI SPLICING
ATTIVAZIONE DI UN SITO CRIPTICO DI SPLICING Attivazione di un sito criptico accettore di splicing nell’introne 1 ccgg SD SA SD SA SA GT AG GT AG ccag

47
MUTAZIONI CHE MODIFICANO SEGNALI IMPORTANTI PER I SITI DI SPLICING
ATTIVAZIONE DI UN SITO CRIPTICO DI SPLICING Attivazione di un sito criptico donatore di splicing nell’esone 2 ccgg SD SA SD SD SA GT AG ccgt GT AG

49
NF2 EXON 4 46561 agaaaattat taacaggctg ctctggcagc cctcattaga acgccgtgag gccatctgtt 46621 gtgatcagcc tacacacctc acttcccctc acagagtatc atgtctccct tgttgctcct 46681 ttcagGTAAA GAAGCAGATT TTAGATGAAA AGATCTACTG CCCTCCTGAG GCTTCTGTGC 46741 TCCTGGCTTC TTACGCCGTC CAGGCCAAGg taggctcaaa gaagaaaaat gtatttttcc 46801 tgggcgttaa tttgggtcat gggatcactc ctatcttctc tttctttggg ttttctgtac 46861 ttcagagaga cttgccccag aaagtgagat gttatgggac ttgtggactt gaagaaccac

50
NF2 EXON 4 46561 agaaaattat taacaggctg ctctggcagc cctcattaga acgccgtgag gccatctgtt 46621 gtgatcagcc tacacacctc acttcccctc acagagtatc atgtctccct tgttgctcct 46681 ttcagGTAAA GAAGCAGATT TTAGATGAAA AGATCTACTG CCCTCCTGAG GCTTCTGTGC 46741 TCCTGGCTTC TTACGCCGTC CAGGCCAAGg taggctcaaa gaagaaaaat gtatttttcc 46801 tgggcgttaa tttgggtcat gggatcactc ctatcttctc tttctttggg ttttctgtac 46861 ttcagagaga cttgccccag aaagtgagat gttatgggac ttgtggactt gaagaaccac

51
RITENZIONE DELL’INTRONE 4
SEQUENZA NORMALE SEQUENZA MUTATA DONOR SITES: POSITION EXON INTRON SCORE GCC GTGAGG CAG GTAAAG . AAA GTGAGA ACCEPTOR SITES: POSITION INTRON EXON SCORE CCCTCACAG AGTA TCCTTTCAG GTAA GCCGTCCAG GCCA GTACTTCAG AGAG DONOR SITES: POSITION EXON INTRON SCORE GCC GTGAGG CAG GTAAAG AAG GTAGGC AAA GTGAGA ACCEPTOR SITES: POSITION INTRON EXON SCORE CCCTCACAG AGTA TCCTTTCAG GTAA GCCGTCCAG GCCA GTACTTCAG AGAG GT AG SD SA RITENZIONE DELL’INTRONE 4

52
MUTAZIONI CHE ALTERANO LO SPLICING
Se gli esoni rimaneggiati non sono in fase: l’omissione dell’esone altera la fase di lettura e portare ad una interruzione prematura della trascrizione. Se gli esoni sono in fase: non viene interrotta la traduzione e si forma una proteina troncata, la cui funzione residua dipende dall’importanza del dominio mancante. ESONE 3 ESONE 4 ATG GAA Met Glu CGA CGT Ala Gly IN FRAME ATG GA Met Glu A CGA CGT Ala Gly NON IN FRAME

54
CLASSE DELLA MUTAZIONE E MODALITA’ IN CUI VIENE ALTERATA L’ESPRESSIONE DEL GENE MUTANTE
Le mutazioni, se cambiano il fenotipo, vengono classificate in due categorie: mutazioni con perdita di funzione “loss of function”; sono per lo più recessive. mutazioni con acquisizione di funzione, “gain of function”; sono molto meno comuni. La mutazione deve essere tale da conferire un’attività anormale alla proteina. Molte sono in sequenze regolatrici.

55
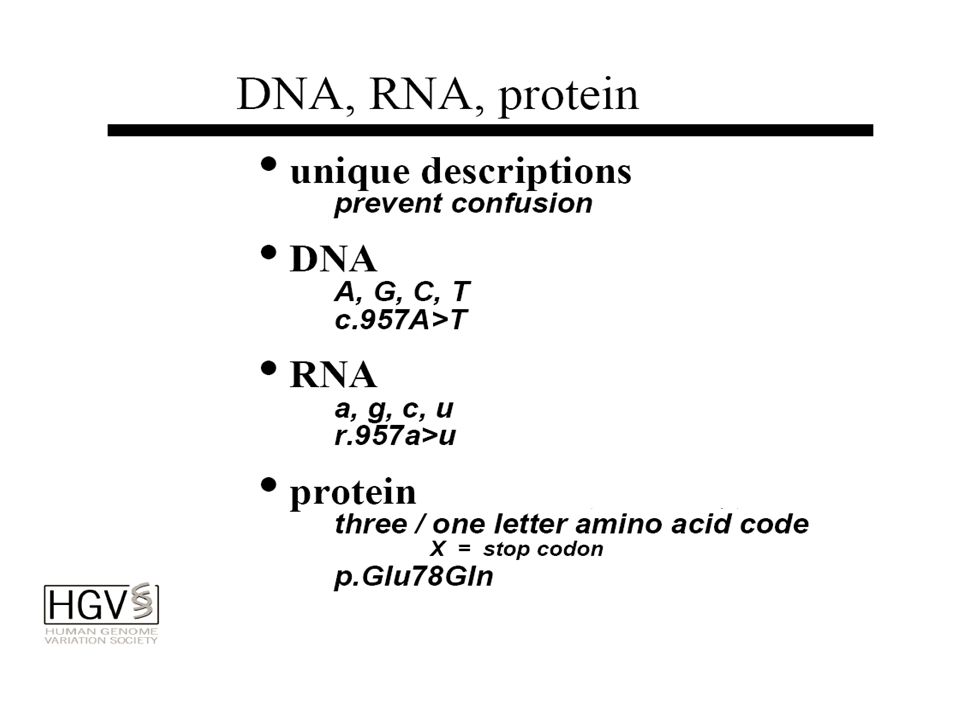
NOMENCLATURA

58
NOMENCLATURA DELLE MUTAZIONI
SOSTITUZIONI NUCLEOTIDICHE La A del codone ATG di inizio è +1; la base immediatamente precedente è -1 Fornire il numero del nucleotide seguito dalla sostituzione: 1162G>A 1997C>T Sostituzioni introniche: specificare il numero dell’introne IVS4+1G>T IVS4-2A>C I I2 I I4 I I6 Ex Ex2 Ex Ex4 Ex Ex6 IVS4+1G>T IVS4-2A>C

59
NOMENCLATURA DELLE MUTAZIONI
DELEZIONI NUCLEOTIDICHE Designate con “del” dopo il numero del nucleotide: 1997delT delTTC INSERZIONI NUCLEOTIDICHE Designate con “ins” dopo il numero dell’intervallo dei nucleotidi interessati insT

60
NOMENCLATURA DELLE MUTAZIONI
BASATA SULL’ALTERAZIONE AMINOACIDICA Codone 1: metionina di inizio Preferibile il codice a singola lettera L’amino acido wild-type viene indicato prima e quello mutato dopo il numero del codone: Y97S (tirosina 97 sostituita da serina) X designa i codoni di stop: R97X (arginina 97 sostituita da stop) Del viene usato per indicare le delezioni di un amino acido: T97del (deleto il codone 97 che codifica per treonina) Ins viene usato per indicare le inserzioni di un amino acido: T97-98ins indica che un codone per treonina è inserito nell’intervallo tra i codoni 97 e 98 della sequenza di riferimento
 X designa i codoni di stop: R97X (arginina 97 sostituita da stop) Del viene usato per indicare le delezioni di un amino acido: T97del (deleto il codone 97 che codifica per treonina) Ins viene usato per indicare le inserzioni di un amino acido: T97-98ins indica che un codone per treonina è inserito nell’intervallo tra i codoni 97 e 98 della sequenza di riferimento.")
61
Questo gruppo di patologie si differenzia
Disordini Genomici Questo gruppo di patologie si differenzia dai disordini mendeliani, in cui il fenotipo è determinato da mutazioni puntiformi

62
RIARRANGIAMENTI STRUTTURALI
Caratteristiche strutturali del genoma umano predispongono al verificarsi di riarrangiamenti cromosomici Sequenze ripetute (dupliconi) rappresentate da: segmenti senza geni frammenti di geni pseudogeni* copie di geni famiglie di geni cluster di geni ripetuti Possono dare origine ad appaiamenti tra sequenze omologhe e successivamente a crossing-over impropri *sequenze di DNA, con struttura analoga a geni espressi, che hanno acquisito una o più mutazioni durante l’evoluzione divenendo incapaci di produrre una proteina
 rappresentate da: segmenti senza geni. frammenti di geni. pseudogeni* copie di geni. famiglie di geni. cluster di geni ripetuti. Possono dare origine ad appaiamenti tra. sequenze omologhe e successivamente. a crossing-over impropri. *sequenze di DNA, con struttura analoga a geni espressi, che hanno acquisito una o più mutazioni durante l’evoluzione divenendo incapaci di produrre una proteina.")
63
Studies on the molecular breakpoints of the recurrent
chromosome rearrangements allowed to understand the causes of structural rearrangements

64
Frequency of recurrent chromosome rearrangements Modified from
Chromosome anomalies Syndrome/disorder Estimated frequency INTERSTITIAL DELETION *del(7)(q11.23q11.23) Williams in20,000-50,000 del(8)(q24.1q2424.1) Langer-Giedion del(11)(p13p13) WAGR in 60, ,000 *del(15)(q12q12) Prader-Willi or Angelman in ,000 *del(17)(p11.2p11.2) Smith-Magenis in 25,000 *del(17)(p12p12) HNPP del(20)(p11.23p11.23) Alagille in 70,000 *del(22)(q11.2q11.2) DiGeoge/velocardiofacial in 4,000 TERMINAL DELETION del(1)(p36.3) Monosomy 1p in 10,000 del(4)(p16) Wolf-Hirschhorn in 50,000 del(5)(p15) Cri-du-chat in 50,000 del(16)(p13.3) Rubinstein-Taybi in 125,000 del(17)(p13.3) Miller-Dieker INTERSTITIAL DUPLICATION dup(17)(p12p13) Russel-Silver *dup(15)(q12q12) Variable features with autism dup(17)(p11.2p11.2) Mild developmental delay *dup(17)(p12p12) Charcot-Marie-Tooth disease type 1A in 2,500 dup(X)(q22q22) Pelizaeus- Merzbacher disease Frequency of recurrent chromosome rearrangements Modified from L. Shaffer and J. Lupski, 2000
(q11.23q11.23) Williams 1 in20,000-50,000. del(8)(q24.1q2424.1) Langer-Giedion del(11)(p13p13) WAGR 1 in 60, ,000. *del(15)(q12q12) Prader-Willi or Angelman 1 in ,000. *del(17)(p11.2p11.2) Smith-Magenis 1 in 25,000. *del(17)(p12p12) HNPP del(20)(p11.23p11.23) Alagille 1 in 70,000. *del(22)(q11.2q11.2) DiGeoge/velocardiofacial 1 in 4,000. TERMINAL DELETION. del(1)(p36.3) Monosomy 1p 1 in 10,000. del(4)(p16) Wolf-Hirschhorn 1 in 50,000. del(5)(p15) Cri-du-chat 1 in 50,000. del(16)(p13.3) Rubinstein-Taybi 1 in 125,000. del(17)(p13.3) Miller-Dieker INTERSTITIAL DUPLICATION. dup(17)(p12p13) Russel-Silver *dup(15)(q12q12) Variable features with autism dup(17)(p11.2p11.2) Mild developmental delay *dup(17)(p12p12) Charcot-Marie-Tooth disease type 1A 1 in 2,500. dup(X)(q22q22) Pelizaeus- Merzbacher disease Frequency of. recurrent. chromosome. rearrangements. Modified from. L. Shaffer. and. J. Lupski,")
65
BASI MOLECOLARI DELLE MUTAZIONI SPONTANEE
Inserzione/delezione di grandi regioni Inserzioni/delezioni estese sono causate da ricombinazione omologa ineguale (sequenze omologhe non alleliche) o ricombinazione non omologa (sequenze non omologhe o solo parzialmente omologhe, intersperse nel genoma.
 o ricombinazione non omologa (sequenze non omologhe o solo parzialmente omologhe, intersperse nel genoma.")
66
TALASSEMIA ALFA - 2 catene alfa e 2 catene beta vanno a costituire l’emoglobina normale; nella talassemia c’è un difetto in una delle due catene; nella talassemia alfa il difetto è nella sintesi delle catene alfa.

67
GENI RIPETUTI IN TANDEM
TALASSEMIA ALFA GENI RIPETUTI IN TANDEM E’ causata per lo più da delezioni del gene α-globina il cluster dei geni α è localizzato in 16p13.3 e

68
LA PERDITA DI UNA COPIA DEL GENE PORTA AD APLOINSUFFICIENZA
TALASSEMIA ALFA I geni α1 e α2 sono quasi identici a livello di sequenza e codificano per proteine identiche: un evento di ricombinazione ineguale induce uno scambio tra cromosomi omologhi con la formazione di un cromosoma con geni α-globinici in più e l’altro con riduzione del numero o assenza dei geni α un numero aumentato di geni del cluster α-globinico non sembra legato a conseguenze cliniche LA PERDITA DI UNA COPIA DEL GENE PORTA AD APLOINSUFFICIENZA

69
Talassemie (diminuzione della sintesi delle catene a o b)
normale La perdita di geni a-globinici porta a forme di a-talassemia di differente severità a2-talassemia (portatore silente) a1-talassemia (anemia non significativa) Hb H disease (anemia da lieve a severa) Idrope fetale (morte fetale o in epoca neonatale)
 a1-talassemia. (anemia non significativa) Hb H disease. (anemia da lieve a severa) Idrope fetale. (morte fetale o in epoca neonatale)")
70
MUTAZIONI DINAMICHE

71
Mutazioni dinamiche e patologia
Malattia localizzazione ripetizione della sequenza espansioni modeste in regioni codificanti: seq. normali ca 4-40 ripetizioni, geni mutati ca ripetizioni Huntington’s disease codificante (CAG)n Spinocerebellar ataxia type 1 codificante (CAG)n (SCA1) Machado-Joseph’s disease codificante (CAG)n Kennedy’s disease (SBMA) codificante (CAG)n Dentatorubral-pallidoluysian codificante (CAG)n espansioni molto estese in regioni non codificanti: seq. normali 2-55 ripetizioni, geni mutati ca ripetizioni Fragile XA syndrome 5’ UTR (CGG)n Myotonic dystrophy 3’ UTR (CTG)n
n. Spinocerebellar ataxia type 1 codificante (CAG)n. (SCA1) Machado-Joseph’s disease codificante (CAG)n. Kennedy’s disease (SBMA) codificante (CAG)n. Dentatorubral-pallidoluysian codificante (CAG)n. espansioni molto estese in regioni non codificanti: seq. normali 2-55 ripetizioni, geni mutati ca ripetizioni. Fragile XA syndrome 5’ UTR (CGG)n. Myotonic dystrophy 3’ UTR (CTG)n.")
72
Nel 1991 sono state scoperte le espansioni instabili di ripetizioni trinucleotidiche che hanno messo in luce un nuovo meccanismo patogenetico. Queste triplette possono inserirsi al 3’ , al 5’, nelle regioni codificanti e non codificanti di un gene e sono responsabili di diversi tipi di patologie Si definiscono ripetizioni instabili perchè il numero delle ripetizioni tende a crescere (espandersi) con l’aumentare delle generazionia causa di crossing-over ineguali alla meiosi
 con l’aumentare delle generazionia causa di crossing-over ineguali alla meiosi.")
73
MALATTIE DA ESPANSIONE DI TRIPLETTE - GENETICA
Sequenze ripetute di trinucleotidi sono presenti in tutto il genoma e sono generalmente trasmesse stabilmente. Si trovano in regioni codificanti o non codificanti di un gene e il loro numero varia da individuo a individuo In alcuni geni se il numero delle triplette supera determinati valori rende la sequenza altamente instabile determinando un aumento delle ripetizioni nelle generazioni successive. L’espansione oltre una determinata soglia può influire sull’espressione genica e sulla stabilità dell’mRNA determinando l’insorgenza di sintomi clinici.

74
MUTAZIONI DINAMICHE in regioni non codificanti

75
MUTAZIONI DINAMICHE in regioni codificanti, associate ad espansione di poliglutamine

76
Malattie associate ad espansioni ripetute di trinucleotidi
Normale 6-55 Mutazione >200 Normale 5-35 Mutazione Normale 7-21 Mutazione Normale 11-35 Mutazione Normale 19-36 Mutazione 43-81 Le espansioni ripetute di trinucleotidi interferiscono con l’espressione del gene o della proteina codificata

77
MALATTIE DA ESPANSIONE DI TRIPLETTE
L’evento primario alla base di queste malattie è l’espansione del numero di triplette . Questo può causare: Inattivazione del gene con soppressione della trascrizione e conseguente mancata produzione della proteina (gene FMRi nella sindrome dell’X fragile) Diminuzione del livello di trascrizione (distrofia miotonica) Produzione di una proteina alterata con perdita di funzione (HD, SCA)
 Diminuzione del livello di trascrizione (distrofia miotonica) Produzione di una proteina alterata con perdita di funzione (HD, SCA)")
78
ANTICIPAZIONE Comparsa più PRECOCE o MAGGIORE GRAVITÀ DEI SINTOMI CLINICI con il passare delle generazioni La gravità di queste patologie correla direttamente con la grandezza dell’espansione (HD,DM) 100 DM 300 400 700 cDM 1000 1400 I, II, II,3 III,1 III,2 III,3 I II III
 100. DM cDM I,1 II,2 II,3 III,1 III,2 III,3. I. II. III")
79
Distrofia Miotonica patologia autosomica dominante
caratterizzata da miotonia e progressiva degenerazione muscolare la forma più comune di distrofia muscolare con esordio nell’adulto manifestazioni scheletriche, cardiovascolari, e oculari (cataratta) associazione con cambiamenti cognitivi, incluso il ritardo mentale la patologia mostra il fenomeno dell’“anticipazione” esordio tardivo con sintomi lievi nella prima generazione quindi esordio neo-natale, associato a ritardo mentale, alla 3°-4° generazione causata da mutazioni nel gene DM-1 (miotonina protein kinasi), espresso nel cervello, cuore, e muscolo il gene normale ha al 3’-UTR una ripetizione CTG polimorfica nella popolazione (5-30 ripetizioni) i pazienti hanno un numero espanso di ripetizioni, fino a molte centinaia, che causano una diminuzione dei livelli di mRNA
 associazione con cambiamenti cognitivi, incluso il ritardo mentale. la patologia mostra il fenomeno dell’ anticipazione esordio tardivo con sintomi lievi nella prima generazione. quindi esordio neo-natale, associato a ritardo mentale, alla 3°-4° generazione. causata da mutazioni nel gene DM-1 (miotonina protein kinasi), espresso nel cervello, cuore, e muscolo. il gene normale ha al 3’-UTR una ripetizione CTG polimorfica nella. popolazione (5-30 ripetizioni) i pazienti hanno un numero espanso di ripetizioni, fino a molte centinaia, che causano una diminuzione dei livelli di mRNA.")
80
Anticipazione la severità della patologia aumenta di generazione in generazione i sintomi passano da lievi nella prima generazione a severi nelle generazioni più avanzate è dovuta all’espansione, a step successivi, delle ripetizioni trinucleotidiche nella popolazione normale, la lunghezza della ripetizione è polimorfica, ma stabile il primo step è la formazione di una “premutazione” con normale fenotipo ma instabile la premutazione quindi si espande nella successiva generazione a una lunghezza molto maggiore e una ulteriore instabilità 100 DM 300 400 700 cDM 1000 1400 I, II, II,3 III,1 III,2 III,3 I II III

81
Structure and inheritance of CTG repeats in myotonic dystrophy
affected >75 premutation 45-75 myotonic protein kinase gene normal 5-30 (CTG)n
n.")
82
Fragile X syndrome (FRAXA)
X-linked Frequenza: 1 maschio ogni ~1.250 è la seconda causa di ritardo mentale La patologia mostra il fenomeno dell’anticipazione Aumenta la “penetranza” nelle generazioni successive Causata da mutazioni nel gene FMR-1, espresso ad alti livelli nel cervello e nei testicoli, e ampiamente nell’embrione Il gene normale ha al 5’-UTR la sequenza CGG ripetuta e polimorfica Nella popolazione (6-55 ripetizioni) I pazienti hanno un numero di ripetizioni che raggiunge le migliaia Ne risulta il silenziamento trascrizionale
 I pazienti hanno un numero di ripetizioni che raggiunge le migliaia. Ne risulta il silenziamento trascrizionale.")
83
Structure and inheritance of CGG repeats in fragile X syndrome
affected >200 premutation normal 6-55 FMR-1 gene (CGG)n
n.")
84
Huntington’s disease Patologia autosomica dominante
Esordio sia giovanile che adulto Associato a movimenti involontari (chorea) Malattia neurodegenerativa ereditaria causata da disturbi del movimento, modificazioni della personalità e demenza con progressiva disabilità. La morte sopravviene in genere entro i anni dall’insorgenza. Causata da mutazioni nel gene IT15 (la cui funzione non è nota) Il gene normale ha una ripetizione CAG polimorfica nella popolazione (11-35 ripetizioni) I pazienti hanno un numero espanso di ripetizioni (>35) La ripetizione CAG è tradotta in tratti di poliglutamina nella proteina La patologia mostra il fenomeno dell’anticipazione, ma la severità non sempre correla strettamente con il grado di espansione
 Malattia neurodegenerativa ereditaria causata da disturbi del movimento, modificazioni della personalità e demenza con progressiva disabilità. La morte sopravviene in genere entro i anni dall’insorgenza. Causata da mutazioni nel gene IT15 (la cui funzione non è nota) Il gene normale ha una ripetizione CAG polimorfica nella popolazione (11-35 ripetizioni) I pazienti hanno un numero espanso di ripetizioni (>35) La ripetizione CAG è tradotta in tratti di poliglutamina nella proteina. La patologia mostra il fenomeno dell’anticipazione, ma la severità non. sempre correla strettamente con il grado di espansione.")
85
MALATTIA AUTOSOMICA DOMINANTE
COREA DI HUNTINGTON MALATTIA AUTOSOMICA DOMINANTE PENETRANZA COMPLETA

86
CAG repeats in Huntington’s disease
affected range: >39 repeats may or may not have disease: repeats normal but can expand: repeats normal range: repeats (CAG)n (Gln)n
n. (Gln)n.")
87
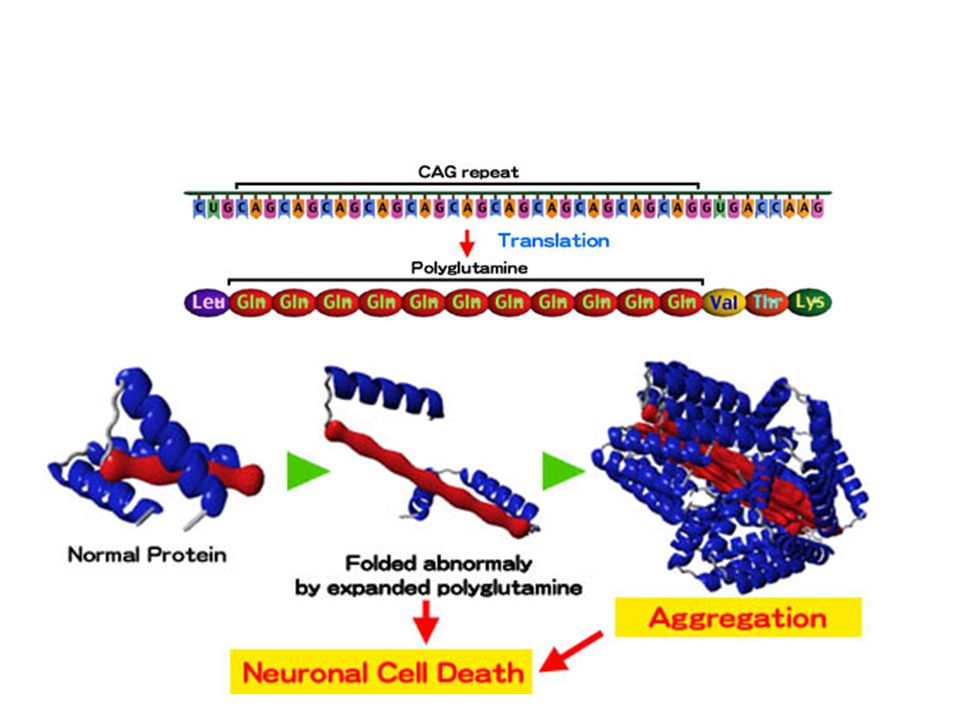
TRATTO POLIGLUTAMINICO
<29 triplette: fenotipo normale 29-34: fenotipo normale, allele instabile 35-39: zona d’ombra; marcata instabilità > 40: patologico, altamente instabile

Presentazioni simili



















 DI BREVI TRATTI RIPETUTI>")


