Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
L’imprinting genomico

2
EPIGENETICA Per epigenetica si intende qualsiasi attività di regolazione dei geni tramite processi chimici che non comportano cambiamenti nella sequenza del DNA ma possono indurre fenotipi diversi nell’individuo o nella progenie Queste modificazioni fanno si che diminuisca il grado di accessibilità del DNA per i fattori di trascrizione alterando l’attività di tale gene.

3
ALTERAZIONE STABILE DELLA POTENZIALE ESPRESSIONE GENICA
……………………… quando avviene ? Avviene durante lo sviluppo e la proliferazione cellulare, senza alterare la sequenza genica sulla quale interviene. Ruolo importante dimostrato in: Biologia del cancro; Infezioni virali; Tecnologie transgeniche; Imprinting

4
CONTROLLO EPIGENETICO DELL’ESPRESSIONE GENETICA
MANTENIMENTO DELL’ESPRESSIONE DIFFERENZIALE DI GENI NEI DIVERSI TESSUTI METILAZIONE DEL DNA ACETILAZIONE DEGLI ISTONI

5
METILAZIONE. È uno degli eventi epigenetici più frequenti nel genoma dei mammiferi. Modificazione chimica covalente EREDITABILE ma REVERSIBILE. Consiste nell’aggiunta di un gruppo metile (-CH3) al carbonio in 5’ dell’anello di una CITOSINA. Nei vertebrati la metilazione interessa esclusivamente la CITOSINA
 al carbonio in 5’ dell’anello di una CITOSINA. Nei vertebrati la metilazione interessa esclusivamente la CITOSINA.")
6
La metilazione del DNA è un processo biochimico mediante il quale l’enzima DNA-metiltransferasi (DNMT) aggiunge un gruppo metilico alle citosine, (se seguite da una guanina) trasformandola in 5-metilcitosina N N DNMT H3C 4 5 3 N N 6 2 1 METILAZIONE N O N O citosina 5-metilcitosina

7
METILAZIONE. Il genoma umano non è metilato uniformemente; contiene regioni non metilate interposte ad altre metilate. La maggior parte della metilazione in C avviene nel contesto della sequenze 5’-CG-3’ dette “CpG island”.

8
Quali regioni sono bersaglio della metilazione?
CpG islands •Piccole regioni IPERMETILATE del DNA di kb ca. •Si ritrovano in media ogni 100 kb. •Nell’uomo circa la metà dei geni (geni house-keeping e geni con pattern d’espressione tessuto-specifica) ha CpG islands.
 ha CpG islands.")
9
“CpG islands” L’aumento della metilazione nella regione promotore di un gene porta ad una ridotta espressione, mentre la metilazione nella regione trascritta ha un effetto variabile

10
IMPRINTING GENOMICO Cos’è l’imprinting?

11
IMPRINTING GENOMICO E’ un meccanismo epigenetico di regolazione trascrizionale attraverso il quale alcuni geni vengono espressi o meno a seconda della loro origine parentale L’esistenza dell’imprinting genomico suggerisce invece che il genoma materno e quello paterno non sono funzionalmente equivalenti, ma hanno un ruolo supplementare e non sostituibile nel determinare un corretto sviluppo embrionale

12
In questo albero, la patologia è sempre trasmessa per via materna
Modified by J Med Genet 29: , 1992 In questo albero, la patologia è sempre trasmessa per via materna e non in modo mendeliano

13
Imprinting paterno Penetranza elevata quando il locus e’ ereditato dalla madre Allele paterno silente

14
Imprinting materno il carattere viene espresso quando ereditato dal padre l’allele materno e’ silente

15
Alcuni geni sottoposti ad imprinting
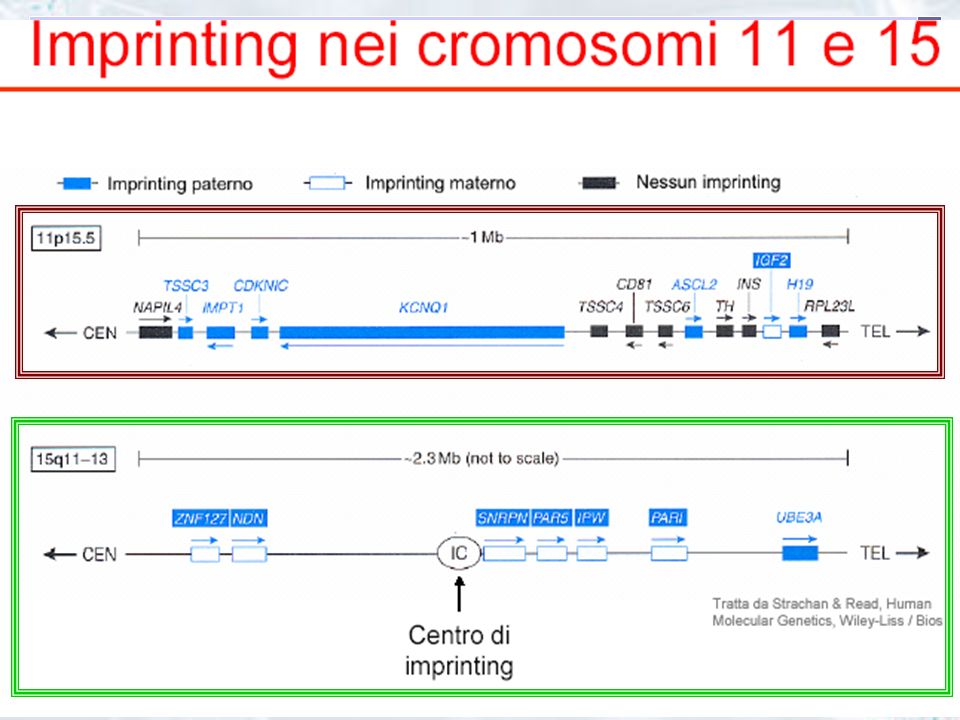
Chr1 (p73); Chr5 (U2AFBPL); Chr6 (MAS1; M6P/IGF2R..); Chr7 (GAMMA2-COP..); Chr11 (H19;IGF2;INS..); Chr13 (HTR2A); Chr14 (MEG3/GTL2); Chr15 (GABRA5;GABRB3;NDN;PAR1;PAR5;SNRPN;UBE3A) Chr18 (IMPACT); Chr19 (PEG3); Chr20 (GNAS1;NEURONATIN) ChrX (XIST)
; Chr5 (U2AFBPL); Chr6 (MAS1; M6P/IGF2R..); Chr7 (GAMMA2-COP..); Chr11 (H19;IGF2;INS..); Chr13 (HTR2A); Chr14 (MEG3/GTL2); Chr15 (GABRA5;GABRB3;NDN;PAR1;PAR5;SNRPN;UBE3A) Chr18 (IMPACT); Chr19 (PEG3); Chr20 (GNAS1;NEURONATIN) ChrX (XIST)")
16
Come e quando? Non e’ un cambiamento permanente del DNA
Deve essere abolito prima di trasmettere il genoma Deve essere ripristinato coerentemente al sesso dell’individuo che trasmette Su questa nuova “etichettatura” agiscono i meccanismi di regolazione genica secondo i pattern previsti per cellule, tessuti.....

17
Determinazione e propagazione dell’imprinting
L’imprinting deve essere mantenuto durante la divisione delle cellule somatiche L’imprinting deve essere cancellabile nella linea germinale per consentire la determinazione del pattern di imprinting del sesso opposto

18
Imprinting e patologia
• Perdita dell’allele non soggetto ad imprinting: assenza di proteina • Perdita dell’imprinting (LOI, loss of imprinting): livello di proteina raddoppiato rispetto al normale
: livello di proteina. raddoppiato rispetto al normale.")
19
Alcuni geni sottoposti ad imprinting
Chr1 (p73); Chr5 (U2AFBPL); Chr6 (MAS1; M6P/IGF2R..); Chr7 (GAMMA2-COP..); Chr11 (H19;IGF2;INS..); Chr13 (HTR2A); Chr14 (MEG3/GTL2); Chr15 (GABRA5;GABRB3;NDN;PAR1;PAR5;SNRPN;UBE3A) Chr18 (IMPACT); Chr19 (PEG3); Chr20 (GNAS1;NEURONATIN) ChrX (XIST)
; Chr5 (U2AFBPL); Chr6 (MAS1; M6P/IGF2R..); Chr7 (GAMMA2-COP..); Chr11 (H19;IGF2;INS..); Chr13 (HTR2A); Chr14 (MEG3/GTL2); Chr15 (GABRA5;GABRB3;NDN;PAR1;PAR5;SNRPN;UBE3A) Chr18 (IMPACT); Chr19 (PEG3); Chr20 (GNAS1;NEURONATIN) ChrX (XIST)")
21
Sindrome di Prader-Willi
Incidenza: 1/ /15.000

22
Sindrome di Prader-Willi: Criteri Diagnostici Maggiori
Ipotonia centrale infantile con suzione scarsa, tendente a migliorare nel tempo Difficoltà di alimentazione e scarso accrescimento Aumento eccessivo di peso dopo 1 a. e prima di 6 a. Anomalie facciali: costrizione bitemporale occhi a mandorla bocca piccola con labbro superiore sottile e angoli rivolti in basso Ipogonadismo Ritardo mentale medio-moderato Iperfagia/ossessione per il cibo Anomalia cromosomica o molecolare della regione 15q11-q13

23
Microdelezione del cromosoma 15 nella sindrome di Prader-Willi
MAT PAT del 15(q11-q13) Visibile con FISH (o con esame citogenetico ad alta risoluzione)
 Visibile con FISH (o con esame citogenetico ad alta risoluzione)")
24
Sindrome di Angelman Incidenza: 1/ /30.000

25
Sindrome di Angelman: Caratteristiche Cliniche Costanti
Grave ritardo di sviluppo psicomotorio Assenza, completa o pressoché completa, del linguaggio Andatura atassica e/o movimenti tremuli Anomalie comportamentali: riso non motivato<< carattere ipereccitabile, con tendenza ad agitare le mani deficit di attenzione atteggiamento apparentemente felice iperattività motoria

26
Sindrome di Angelman: Caratteristiche Cliniche Frequenti (>80%)
Microcefalia prima dei 2 anni Convulsioni in genere <3 anni Pattern EEG anormale, con ampie onde lente, la cui comparsa è facilitata dalla chiusura degli occhi

27
PERDITA DELL’IMPRINTING
1) Alterazioni di metilazione :eccesso o riduzione 2) Presenza di 2 copie dello stesso allele (DISOMIA UNIPARENTERALE)
 Alterazioni di metilazione :eccesso o. riduzione. 2) Presenza di 2 copie dello stesso allele. (DISOMIA UNIPARENTERALE)")
28
Soggetto normale AS PWS Delezione 15q11-q13: 70%

29
Nei rimanenti casi il cariotipo è, in genere, normale
La FISH con una sonda specifica del centromero del 15 ed una della regione PWS/AS identifica una delezione eterozigote (paterna in PWS e materna in AS)alla regione critica nel 70% dei casi Nei rimanenti casi il cariotipo è, in genere, normale
alla regione critica nel 70% dei casi. Nei rimanenti casi il cariotipo è, in genere, normale.")
30
PERDITA DELL’IMPRINTING
1) Alterazioni di metilazione :eccesso o riduzione 2) Presenza di 2 copie dello stesso allele (DISOMIA UNIPARENTERALE)
 Alterazioni di metilazione :eccesso o. riduzione. 2) Presenza di 2 copie dello stesso allele. (DISOMIA UNIPARENTERALE)")
31
PWS Delezione 15q11-q13: 70% Mat UPD:25% Soggetto normale AS
Pat UPD:2% Mat UPD:25%

33
46 cromos 24 cromos 22 cromos 22 cromat 24 cromat Non disgiunz. alla MI

34
Uovo con nullisomia per il cromosoma 15
45 cromosomi: monosomia 15 duplicaz. selettiva 15: 46 cromosomi con UPD 15 paterno

35
Le sindromi di Angelmann e di Prader-Willi
sono esempi di patologie dovute all’imprinting genomico Sindrome di Prader-Willi Sindrome di Angelman

36
AS PWS 15 der(15) 22 der(22) 15 der(22) 22 The Lancet, 338:638 (1991)
 22 der(22) 15 der(22) 22 The Lancet, 338:638 (1991)")
37
(sindrome di Angelman o sindrome di Prader-Willi)
La stessa traslocazione sbilanciata t(15;22)(q12;q11) è associata a due fenotipi diversi (sindrome di Angelman o sindrome di Prader-Willi) a seconda che sia ereditata dalla madre anziché dal padre
(q12;q11) è associata a due fenotipi diversi. (sindrome di Angelman o sindrome di Prader-Willi) a seconda che sia ereditata dalla madre anziché dal padre.")
38
IMPRINTING GENOMICO: MECCANISMO MOLECOLARE
Metilazione genica, a livello delle sequenze CpG blocco della trascrizione I geni delle regioni soggette ad imprinting sono particolarmente ricchi di isole CpG cg cg cg cg cg ccggcg regione PWS nel cromosoma 15 paterno CH3 CH3 CH3 CH3 CH3 CH3 CH3 cg cg cg cg cg ccggcg regione PWS nel cromosoma 15 materno

39
Struttura della regione
15q11 Cromosoma 15

40
DIFETTI GENETICI NELLA SINDROME DI PRADER-WILLI
crom. 15 paterno X crom. 15 materno 1. Microdelezione regione PWS cromosoma 15 paterno: 70% Evento sporadico, rischio di ricorrenza trascurabile crom. 15 materno 2. Disomia uniparentale materna o riarrangiamenti cromosoma 15: 25% Correlata con età materna Evento sporadico, rischio di ricorrenza trascurabile crom. 15 paterno crom. 15 materno 3. Difetti del Centro dell’Imprinting cromosoma paterno: <5% Rischio di ricorrenza 50% se padre portatore di microdelezione IC

41
Alcuni casi di sindrome di Angelman sono familiari!
Mutazioni puntiformi inattivanti (“loss of function”) del gene UBE3A UBE3A è contenuto nella regione AS ed è soggetto ad imprinting paterno Prodotto proteico: ubiquitin-ligasi enzima che lega proteine destinate alla degradazione all’ubiquitina Forma autosomica dominante, con espressione dipendente dal sesso del genitore trasmettitore madre trasmettitrice s. di Angelman se trasmesso allele mutato padre trasmettitore nessuna conseguenza
 del gene UBE3A. UBE3A è contenuto nella regione AS ed è soggetto ad imprinting paterno. Prodotto proteico: ubiquitin-ligasi enzima che lega proteine destinate alla degradazione all’ubiquitina. Forma autosomica dominante, con espressione dipendente dal sesso del genitore trasmettitore. madre trasmettitrice s. di Angelman se trasmesso allele mutato. padre trasmettitore nessuna conseguenza.")
42
Effetto di origine parentale nella sindrome di Angelman dovuta a mutazioni di UBE3A
wt mat. att. mut. pat. inatt. wt mat. att. wt pat. inatt. wt mat. att. mut. pat. inatt. wt mat. att. mut. pat. inatt. wt mat. att. wt pat. inatt. wt mat. att. mut. pat. inatt. wt pat. inatt. mut. mat. inatt. wt pat. inatt. mut. mat. inatt. wt mat. att. mut. pat. inatt. wt mat. att. wt pat. inatt. wt mat. att. mut. pat. inatt. wt mat. att. wt pat. inatt. wt mat. att. wt pat. inatt. wt pat. inatt. mut. mat. inatt.

43
DIFETTI GENETICI NELLA SINDROME DI ANGELMAN
crom. 15 materno X crom. 15 paterno 1. Microdelezione regione AS cromosoma 15 materno: 70% UBE3A crom. 15 paterno 2. Disomia uniparentale paterna cromosoma 15: 2-3% UBE3A crom. 15 materno 3. Difetti Centro dell’Imprinting cromosoma 15 materno: 3-5% UBE3A crom. 15 materno crom. 15 paterno 4. Mutazione puntiforme UBE3A cromosoma materno: 5-7% UBE3A 5. Altri meccanismi (sconosciuti): 12-18%
: 12-18%")
44
La sindrome di Beckwith–Wiedemann
1: nati vivi La Sindrome di Beckwith-Wiedemann è una sindrome da iperaccrescimento Dimostrato il linkage con 11p15 e l’alterazione dell’imprinting genomico di geni presenti in questa regione La maggior parte dei casi sono sporadici ma una piccolo numero sono familiari con trasmissione AD Coinvolgimento della deregolazione dell’imprinting genomico nella regione 11p15 Presenta disordini clinici eterogenei

45
La sindrome di Beckwith–Wiedemann caratteristiche cliniche
Macroglossia Difetti della parete addominale Visceromegalia(macrosomia) Gigantismo natale e postatale Anomalie alle orecchie (fossette, pieghe ecc) Ipoglicemia neonatale Citomegalia e cisti della corteccia surrenale Nefromegalia e anomalie strutturali del rene Emiperplasia (crescita asimmetrica di una o più regioni del corpo) Dismorfismi facciali Palatoschisi Occipite prominente
 Gigantismo natale e postatale Anomalie alle orecchie (fossette, pieghe ecc) Ipoglicemia neonatale. Citomegalia e cisti della corteccia surrenale. Nefromegalia e anomalie strutturali del rene. Emiperplasia (crescita asimmetrica di una o più regioni del corpo) Dismorfismi facciali. Palatoschisi. Occipite prominente.")
46
La sindrome di Beckwith–Wiedemann
Perdita di udito (conduttiva o neurosensoriale) Tonsille e adenoidi di dimensioni notevoli Scoliosi (dovuta all’asimmetria) Tumore di Wilms Epatoblastoma Neuroblastoma Nefroblastoma Tumori delle ghiandole surrenali Tumore del pancreas
 Tonsille e adenoidi di dimensioni notevoli Scoliosi (dovuta all’asimmetria) Tumore di Wilms Epatoblastoma Neuroblastoma Nefroblastoma Tumori delle ghiandole surrenali Tumore del pancreas.")
47
GENI “ IMPRINTED”-BWS Il cluster di geni “imprinted”nella regione 11p15 contiene almeno 12 geni in due distinti domini regolati in cis da due imprinting centers (DMRs)
")
48
DOMINIO 1 H19 codifica un trascritto polII non tradotto GF fetale–espressione paterna Al 3’ del gene H19 un set di enhancers IGF2 e H19 competono per tali enhancers DMR1 (2 kb a monte di H19): Regola l’espressione di IGF2 e H19 Rosso: geni ad espressione materna Blu: geni ad espressione paterna BWS causata dall’aumentato livello d’espressione del gene IGF2 Duplicazione del cromosoma paterno 11p15 Disomia uniparentale paterna Alterazione della metilazione
: Regola l’espressione di IGF2 e H19. Rosso: geni ad espressione materna. Blu: geni ad espressione paterna. BWS. causata dall’aumentato livello d’espressione del gene IGF2. Duplicazione del cromosoma paterno 11p15. Disomia uniparentale paterna. Alterazione della metilazione.")
49
Dominio 2 6 geni imprinted : CDKN1C,TSSC3, SLC22A1L, KCNQ1,TSSC4,PHEMX
KCNQ1OTI: trascritto antisenso che regola negativamente l’espressione dei geni paterni CH3 DMR2 PAT MAT CDKN1C Espressione materna Cdk inibitore Regola negativamente la proliferazione cellulare Mutazioni CDKN1C causano BWS e sono spesso associate con l’eredita autosomica dominante della sindrome SLC22A1L Espressione materna Trasportatore di cationi Mutazioni del gene associata tumore al seno KCNQ1 Espressione materna 6 traslocazoni note sono associate con la BWS L’introne 10 contiene un DMR2

50
Alterazioni genetiche associate a BWS :
DMR2 non metilato con conseguente perdita di imprintig del KCNQ1OTI nel 50% dei casi Disomia uniparentale paterna dell’11p15 nel 10-20% dei casi Perdita di imprintig del gene IGF2 sull’allele materno % dei casi

Presentazioni simili






, L Garagnani (2), L Schirosi (3), C De Gaetani (4), A Maiorana.>")










>")



