Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
La predizione della struttura genica e lo splicing alternativo
Raffaella Rizzi DISCo – Università di Milano-Bicocca

2
Introduzione biologica
DNA Doppia catena polinucleotidica definita sull’alfabeto: A, C, G, T Gene Regione di DNA che codifica proteine

3
Il sequenziamento Cosa significa sequenziare?
determinare la struttura primaria delle molecole biologiche (DNA/RNA e proteine) sequenza dei nucleotidi {a,c,g,t|u} per DNA/RNA g c t Cosa significa sequenziare in generale? Semplicemente ottenere, data una molecola biologica (DNA, RNA o proteina), la sua struttura primaria, ovvero nel caso di DNA/RNA la successione dei nucleotidi. Quindi dal punto di vista informatico il prodotto del sequenziamento di un DNA è una stringa di caratteri definita sull’alfabeto {a,c,g,t}. Poi per un RNA il simbolo ‘t’ (timina) viene sostituito dal simbolo ‘u’ (uracile). Nel caso di una proteina si ottiene una stringa definita sull’alfabeto dei 20 amminoacidi. In questa sede verranno ignorate le proteine perché mi concentrerò solo sugli ambiti della genomica e della trascrittomica e quindi, trovandoci a monte della fase della traduzione in proteine, le sequenze che ci interessano sono di nucleotidi. Più precisamente DNA genomico e trascritti (quindi RNA). ordine degli amminoacidi per le proteine Glu Ile Phe Thr Val His
 sequenza dei nucleotidi {a,c,g,t|u} per DNA/RNA. g. c. t. Cosa significa sequenziare in generale Semplicemente ottenere, data una molecola biologica (DNA, RNA o proteina), la sua struttura primaria, ovvero nel caso di DNA/RNA la successione dei nucleotidi. Quindi dal punto di vista informatico il prodotto del sequenziamento di un DNA è una stringa di caratteri definita sull’alfabeto {a,c,g,t}. Poi per un RNA il simbolo ‘t’ (timina) viene sostituito dal simbolo ‘u’ (uracile). Nel caso di una proteina si ottiene una stringa definita sull’alfabeto dei 20 amminoacidi. In questa sede verranno ignorate le proteine perché mi concentrerò solo sugli ambiti della genomica e della trascrittomica e quindi, trovandoci a monte della fase della traduzione in proteine, le sequenze che ci interessano sono di nucleotidi. Più precisamente DNA genomico e trascritti (quindi RNA). ordine degli amminoacidi per le proteine. Glu. Ile. Phe. Thr. Val. His.")
4
acgttgtgcagtgacggtaa
Il sequenziamento Cosa si ottiene? single-end read DNA/RNA acgttgtgcagtgacggtaa Più concretamente, cosa si ottiene dal sequenziamento di un DNA/RNA… Fondalmentalmente, due tipi di dato. (1) Il cosiddetto single-end read, che è in sostanza un frammento, cioè una sottostringa (un pezzo) di un DNA/RNA.
 Il cosiddetto single-end read, che è in sostanza un frammento, cioè una sottostringa (un pezzo) di un DNA/RNA.")
5
Il sequenziamento Cosa si ottiene dal sequenziamento di una molecola di DNA/RNA paired-end read (o mate-pair) insertion size A B DNA/RNA Oppure… (2) Il cosiddetto paired-end read (chiamato anche mait-pair) che viene ottenuto nel modo seguente. Si estrae praticamente un frammento di una certa dimensione che è nota (e che è chiamata insertion size). Si circolarizza il frammento (i due estremi A e B vengono uniti). Si sequenzia poi una certa porzione a cavallo del punto di congiunzione. Il prodotto è quindi una coppia di frammenti e quindi due stringhe di nucleotidi di lunghezza nota tra le quali esiste una precisa relazione: cioè la loro distanza (insertion size) pensata sulla molecola di provenienza è nota. In genere dato un paired-end read si usa associare ai due frammenti un’orientazione (ad esempio il frammento a sinistra è orientato verso destra e quello a destra è orientato verso sinistra). Lo scopo dell’orientazione serve quando i paired end vengono usati e vengono mappati ad una sequenza di riferimento. A = B insertion size agttgcgt aatgcctg A B
 Il cosiddetto paired-end read (chiamato anche mait-pair) che viene ottenuto nel modo seguente. Si estrae praticamente un frammento di una certa dimensione che è nota (e che è chiamata insertion size). Si circolarizza il frammento (i due estremi A e B vengono uniti). Si sequenzia poi una certa porzione a cavallo del punto di congiunzione. Il prodotto è quindi una coppia di frammenti e quindi due stringhe di nucleotidi di lunghezza nota tra le quali esiste una precisa relazione: cioè la loro distanza (insertion size) pensata sulla molecola di provenienza è nota. In genere dato un paired-end read si usa associare ai due frammenti un’orientazione (ad esempio il frammento a sinistra è orientato verso destra e quello a destra è orientato verso sinistra). Lo scopo dell’orientazione serve quando i paired end vengono usati e vengono mappati ad una sequenza di riferimento. A = B. insertion size. agttgcgt. aatgcctg. A. B.")
6
Il sequenziamento Perché è importante?
… per determinare il genoma di un organismo vivente (problema del Genome Assembly) Genoma Perchè il sequenziamento è importante? Il sequenziamento ha innanzitutto permesso di giungere alla determinazione della intera sequenza del genoma umano (Human Genome Project) e di altri organismi viventi. E il problema cruciale la cui soluzione ha permesso di ottenere la sequenza del genoma è quello di Genome Assembly. Cioè, in input si ha un set di frammenti di DNA (single-end reads) provenienti dal genoma che si vuole determinare (chiaramente questi frammenti devono coprire l’intero genoma, altrimenti manca l’informazione necessaria), essi devono essere assemblati in maniera da ricostruire l’intera sequenza genomica di provenienza
 Genoma. Perchè il sequenziamento è importante Il sequenziamento ha innanzitutto permesso di giungere alla determinazione della intera sequenza del genoma umano (Human Genome Project) e di altri organismi viventi. E il problema cruciale la cui soluzione ha permesso di ottenere la sequenza del genoma è quello di Genome Assembly. Cioè, in input si ha un set di frammenti di DNA (single-end reads) provenienti dal genoma che si vuole determinare (chiaramente questi frammenti devono coprire l’intero genoma, altrimenti manca l’informazione necessaria), essi devono essere assemblati in maniera da ricostruire l’intera sequenza genomica di provenienza.")
7
Sanger Sequencing (1977) Metodo di sequenziamento capillare
Basato su enzima Piuttosto costoso Processa pochissimi reads in un run (un centinaio) Lunghezza frammenti fino a 1000 bp Errore basso Il più importante e diffuso metodo di sequenziamento è stato per più di 30 anni il metodo ideato da Sanger nel E’ un metodo di sequenziamento capillare basato su enzima. Il difetto più evidente è che è molto costo sia in termini di piattaforma di sequenziamento (strumentazioni) che in termini di esperimento di sequenziamento (materiali utilizzati nel protocollo di sequenziamento). Purtroppo non è molto parallelo, in quanto il numero di reads processati in un run è solo di 96. Gli esperimenti richiedono tanto tempo. Il pregio più evidente invece è la qualità del dato prodotto sia in termini di lunghezza dei reads che in termini di errore della sequenza letta (mismatch, delezione e inserimenti).
 Lunghezza frammenti fino a 1000 bp. Errore basso. Il più importante e diffuso metodo di sequenziamento è stato per più di 30 anni il metodo ideato da Sanger nel E’ un metodo di sequenziamento capillare basato su enzima. Il difetto più evidente è che è molto costo sia in termini di piattaforma di sequenziamento (strumentazioni) che in termini di esperimento di sequenziamento (materiali utilizzati nel protocollo di sequenziamento). Purtroppo non è molto parallelo, in quanto il numero di reads processati in un run è solo di 96. Gli esperimenti richiedono tanto tempo. Il pregio più evidente invece è la qualità del dato prodotto sia in termini di lunghezza dei reads che in termini di errore della sequenza letta (mismatch, delezione e inserimenti).")
8
Espressione di un gene tag taa tga [stop] atg…………[stop] 5’ 3’ DNA 3’
TRANSCRIPTION 5’ 3’ exon 1 exon 2 exon 3 pre-mRNA SPLICING by spliceosome CDS atg…………[stop] [stop] tag taa tga splicing product mRNA exon 1 exon 2 exon 3
![Espressione di un gene tag taa tga [stop] atg…………[stop] 5’ 3’ DNA 3’](http://slideplayer.it/slide/578492/2/images/8/Espressione+di+un+gene+tag+taa+tga+%5Bstop%5D+atg%E2%80%A6%E2%80%A6%E2%80%A6%E2%80%A6%5Bstop%5D+5%E2%80%99+3%E2%80%99+DNA+3%E2%80%99.jpg "TRANSCRIPTION. 5’ 3’ exon 1. exon 2. exon 3. pre-mRNA. SPLICING by spliceosome. CDS. atg…………[stop] [stop] tag. taa. tga. splicing product. mRNA. exon 1. exon 2. exon 3.")
9
Pattern di un introne Introni canonici: 99.24 % GT AG
Introni non canonici: 0.69 % 0.05 % 0.02 % GC AG AT AC ALTRO Burset et al., Nucleic Acids Res. 2000, 28:

10
Ma le cose funzionano davvero così?
Espressione di un gene Ma le cose funzionano davvero così? Numero Geni corpo umano : circa Numero Proteine : centinaia di migliaia La corrispondenza 1 a 1 non è rispettata. Perché? SPLICING ALTERNATIVO

11
Alternative Splicing (AS)
Gene 5’ exon 1 exon 2 exon 3 3’ mRNA1 exon 1 exon 2 exon 3

12
Alternative Splicing (AS)
Gene 5’ exon 1 exon 2 exon 3 3’ mRNA1 exon 1 exon 2 exon 3 mRNA2 exon 1’ exon 1’’ exon 2 exon 3

13
Alternative Splicing (AS)
Gene 5’ exon 1 exon 2 exon 3 3’ mRNA1 exon 1 exon 2 exon 3 mRNA2 exon 1’ exon 1’’ exon 2 exon 3 mRNA3 exon 1 exon 3

14
Alternative Splicing (AS)
Gene 5’ exon 1 exon 2 exon 3 3’ mRNA1 exon 1 exon 2 exon 3 mRNA2 exon 1’ exon 1’’ exon 2 exon 3 mRNA3 exon 1 exon 3 mRNA4 exon 1’’’ exon 2 exon 3

15
Alternative Splicing (AS)
Gene 5’ exon 1 exon 2 exon 3 3’ mRNA1 exon 1 exon 2 exon 3 mRNA2 exon 1’ exon 1’’ exon 2 exon 3 mRNA3 exon 1 exon 3 mRNA4 exon 1’’’ exon 2 exon 3 mRNA5 exon 1 exon 2 exon 3’

16
Alternative Splicing (AS)
Gene 5’ exon 1 exon 2 exon 3 exon 4 3’ mRNA1 exon 1 exon 2 exon 4 mRNA2 exon 1 exon 3 exon 4

18
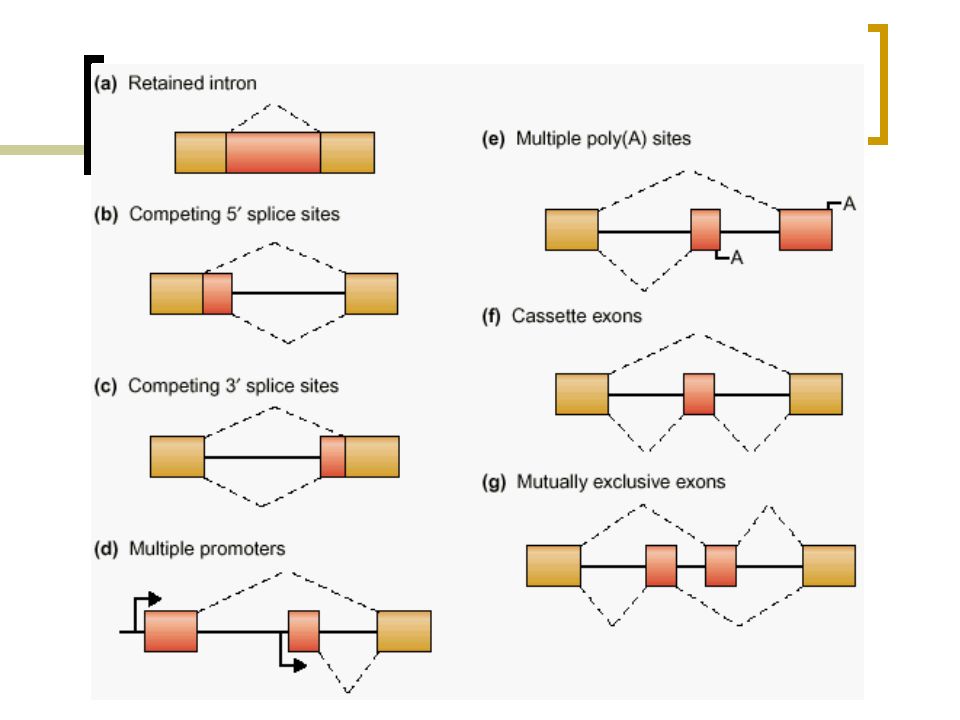
Perché AS è importante? AS avviene nel 40-60% dei geni umani (Modrek and Lee, 2002) AS genera numerosi trascritti a partire da un singolo gene AS is specifico del tessuto in cui si trova la cellula (Graveley, 2001) AS è correlato alle malattie
 AS è correlato alle malattie.")
19
Problema di AS predire le forme di splicing alternativo di un gene
AS è ancora un problema aperto Si ha la necessità di software tools per predire le forme di splicing alternativo di un gene analizzare il meccanismo di splicing tramite la rappresentazione delle possibili isoforme

20
Trascritti e sequenze EST
Un trascritto è l’elenco delle basi (A,T,C,G) che compongono un mRNA maturo Un EST è un frammento di cDNA (copia complementare di un mRNA, prodotta in vitro ) ATTGCGTTAACTGGACTGA mRNA AATTGACCT EST TAACGCAATTGACCTGACT cDNA
 che compongono un mRNA maturo. Un EST è un frammento di cDNA (copia complementare di un mRNA, prodotta in vitro ) ATTGCGTTAACTGGACTGA. mRNA. AATTGACCT. EST. TAACGCAATTGACCTGACT. cDNA.")
21
Expressed Sequence Tag
Cos’è un single-end read da un mRNA (messenger RNA)? EST (Expressed Sequence Tag) Gene C D A B esoni (codificanti) introni (non codificanti) B’ In particolare, se la molecola è un RNA messaggero si ottengo le cosiddette EST (Expressed Sequence Tag). Allora diciamo cos’è un gene e come funziona… Un gene è fondamentalmente una regione del genoma di un organismo che codifica proteine. E’ organizzato in un’alternanza di regioni codificanti (in blu) e regioni non codificanti (in marrone); il confine esone-introne è la giunzione di splicing. Il prodotto dell’espressione di un gene è l’RNA messaggero (o trascritto) che è dato dalla concatenazione di un sottoinsieme dei suoi esoni o di parti di essi. La regola è che l’ordine degli esoni lungo il gene va rispettato. L’mRNA viene poi tradotto in proteina. Inoltre, uno stesso gene può produrre più di un mRNA in dipendenza delle condizioni in cui si trova (stato di salute della cellula, stadio di sviluppo, tessuto, etc.). E quindi può esprimere diverse proteine. Ad esempio per questo gene potremmo avere i tre mRNA che ho disegnato, in cui il primo e il secondo combinano solo tre dei quattro esoni del gene e il terzo addirittura include un prefisso dell’esone B. I diversi mRNA che un gene può esprimere prendono il nome di isoforme. A questo punto viene facile dire cos’è una EST, semplicemente un frammento di mRNA e quindi a meno di troncamento ai bordi e di errori di sequenziamento, una EST è una concatenazioni di regioni codificanti. D A B C B’ mRNA1 mRNA2 mRNA3 EST
 EST (Expressed Sequence Tag) Gene. C. D. A. B. esoni (codificanti) introni (non codificanti) B’ In particolare, se la molecola è un RNA messaggero si ottengo le cosiddette EST (Expressed Sequence Tag). Allora diciamo cos’è un gene e come funziona… Un gene è fondamentalmente una regione del genoma di un organismo che codifica proteine. E’ organizzato in un’alternanza di regioni codificanti (in blu) e regioni non codificanti (in marrone); il confine esone-introne è la giunzione di splicing. Il prodotto dell’espressione di un gene è l’RNA messaggero (o trascritto) che è dato dalla concatenazione di un sottoinsieme dei suoi esoni o di parti di essi. La regola è che l’ordine degli esoni lungo il gene va rispettato. L’mRNA viene poi tradotto in proteina. Inoltre, uno stesso gene può produrre più di un mRNA in dipendenza delle condizioni in cui si trova (stato di salute della cellula, stadio di sviluppo, tessuto, etc.). E quindi può esprimere diverse proteine. Ad esempio per questo gene potremmo avere i tre mRNA che ho disegnato, in cui il primo e il secondo combinano solo tre dei quattro esoni del gene e il terzo addirittura include un prefisso dell’esone B. I diversi mRNA che un gene può esprimere prendono il nome di isoforme. A questo punto viene facile dire cos’è una EST, semplicemente un frammento di mRNA e quindi a meno di troncamento ai bordi e di errori di sequenziamento, una EST è una concatenazioni di regioni codificanti. D. A. B. C. B’ mRNA1. mRNA2. mRNA3. EST.")
22
ESTs Because of their nature, EST sequences are a valuable source of data. They are publicly available through data banks. For example. Unigene, that is accessible via web from the NCBI’s site, stores ESTs grouped by organism and gene. ESTs are mainly used for identifying genes on a genome, for the prediction of the exon-intron structure of a gene and of the alternative transcripts that the gene may potentially express. Le sequenze EST sono dati importanti e disponibili pubblicamente per: identificare geni lungo un genoma predire la struttura in esoni e introni di un geni …e le sue isoforme alternative (alternative splicing prediction) per studi di espressione genica Reference The Unigene Database:
 per studi di espressione genica. Reference. The Unigene Database:")
23
Il sequenziamento di EST
… per determinare la struttura e l’espressione di un gene Perché è importante? Gene C D A B A B A’ C D’ C’ D A’: suffisso di A C’: prefisso di C D’: prefisso di D EST Un altro ambito in cui il sequenziamento è fondamentale è quello della predizione della struttura di un gene e delle proteine che può esprimere. Quindi mappando (allineando) un set di EST alla porzione di genoma relativo ad un gene, si può risalire alla struttura in esoni-introni del gene e agli mRNA che il gene potenzialmente trascrive.
 un set di EST alla porzione di genoma relativo ad un gene, si può risalire alla struttura in esoni-introni del gene e agli mRNA che il gene potenzialmente trascrive.")
24
Splice junctions de-novo
Gene D A’ C B A EST C’ B D’ A’ B Dire che l’uso di SR per le junctions avviene con ss noti. Ora però si incomincia a determinarle ab initio. Però il problema con SR è che non coprono tanti esoni (anzi a volte è uno solo) e la giunzione si può trovare vicina ad un bordo (e ci sono gli errori). A’ B C
 e la giunzione si può trovare vicina ad un bordo (e ci sono gli errori). A’ B. C.")
25
Splice junctions de-novo
Gene C D A B splice junction B A’ C D’ C’ D EST A

26
Perché predire AS è difficile?
La predizione della struttura di un gene è un compito difficile a causa di gli errori di sequenziamento nelle EST rendono difficoltosa la localizzazione delle splice junctions le duplicazioni possono produrre più di un possibile allineamento EST-genomica I dati in input sono enormi: efficienza in tempo e spazio

27
What is available? Fast Blast-like programs to produce a single EST alignment to a genomic sequence (Blat, Sim4): - Spidey (Wheelan et al., 2001) - Squall (Ogasawara & Morishita, 2002) - Ecgene (Kim et al., 2005) - AceView ( - Splicing graphs (Heber et al., 2002)
 - Squall (Ogasawara & Morishita, 2002) - Ecgene (Kim et al., 2005) - AceView ( - Splicing graphs (Heber et al., 2002)")
28
What is available? Fast Blast-like programs to produce a single EST alignment to a genomic sequence (Blat, Sim4): drawbacks - Spidey => independent single EST alignment - Squall => independent single EST alignment - Ecgene => detects variants of more than 15bp - AceView =>over-prediction - Splicing graphs => over-prediction

29
ASPIC software ASPIC (Alternative Splicing PredICtion) implements an optimization strategy that: performs a multiple alignment of transcript data to the genomic sequence detects the intron set that minimizes the number of splicing sites generates the minimal set of transcript isoforms compatible with the detected splicing events P. Bonizzoni, R. Rizzi, G. Pesole. ASPIC: a novel method to predict the exon-intron structure of a gene that is optimally compatible to a set of transcript sequences. BMC Bioinformatics (2005), 6(1):244. T. Castrignanò, R. Rizzi, I.G. Talamo, P. D’Onorio De Meo, A. Anselmo, P. Bonizzoni, G. Pesole. ASPIC: a web resource for alternative splicing prediction and transcript isoforms characterization. Nucleic Acids Research (2006), 34(Web Server Issue):W440:3.
, 6(1):244. T. Castrignanò, R. Rizzi, I.G. Talamo, P. D’Onorio De Meo, A. Anselmo, P. Bonizzoni, G. Pesole. ASPIC: a web resource for alternative splicing prediction and transcript isoforms characterization. Nucleic Acids Research (2006), 34(Web Server Issue):W440:3.")
30
ASPIC web site http://www.caspur.it/ASPIC/ INPUT: A genomic sequence
by pasting a sequence into a text box by uploading a sequence as a text file by specifying an ENSEMBL ID or a HUGO name (only for human) by specifying a chromosomal range
 by specifying a chromosomal range.")
31
ASPIC web site http://www.caspur.it/ASPIC/ INPUT:
A collection of transcripts by pasting them into a text box by uploading them as a text file by specifying a UNIGENE ID The minimum dimension for exons on the genomic sequence

32
ASPicDB ASPicDB (http://www.caspur.it/ASPicDB/index.php)
database of alternative splicing annotations of human genes ASPicDB was obtained by ASPic algorithm Funded by MIUR - FIRB LIBI 32 32

33
ASPicDB

34
ASPicDB

35
Ricostruzione isoforme
Problema di ricostruzione di isoforme full-length Data la struttura in exoni-introni del gene, trovare tutte le possibili combinazioni di esoni (o parti di essi) che danno luogo ad un isoforma completa Gene C D A B C D A B Isoform#1 Isoform#2 Isoform#3
 che danno. luogo ad un isoforma completa. Gene. C. D. A. B. C. D. A. B. Isoform#1. Isoform#2. Isoform#3.")
Presentazioni simili


>")


 & SQL Server 2005 Express.>")













>")