Scaricare la presentazione
1
anemia sanguinamento (acuto/cronico) iporigenerativa aumentata distruzione (emolitica) progenitori eritroidi (anemia aplastica, CDA, PRCA) sintesi Hb (carenza Fe, talassemia) sintesi di DNA (carenza B12, ac folico) anemia da disordine cronico infiltrazione midollare (mieloftisi) anemia sideroblastica difetto intrinseco difetto estrinseco AEA agenti fisici o chimici infezioni farmaci ipersplenismo difetti di membrana difetti enzimatici porifirie emoglobinopatie PNH
 iporigenerativa aumentata distruzione (emolitica) progenitori eritroidi (anemia aplastica, CDA, PRCA) sintesi Hb (carenza Fe, talassemia) sintesi di DNA (carenza B12, ac folico) anemia da disordine cronico infiltrazione midollare (mieloftisi) anemia sideroblastica difetto intrinseco difetto estrinseco AEA agenti fisici o chimici infezioni farmaci ipersplenismo difetti di membrana difetti enzimatici porifirie emoglobinopatie PNH")
2
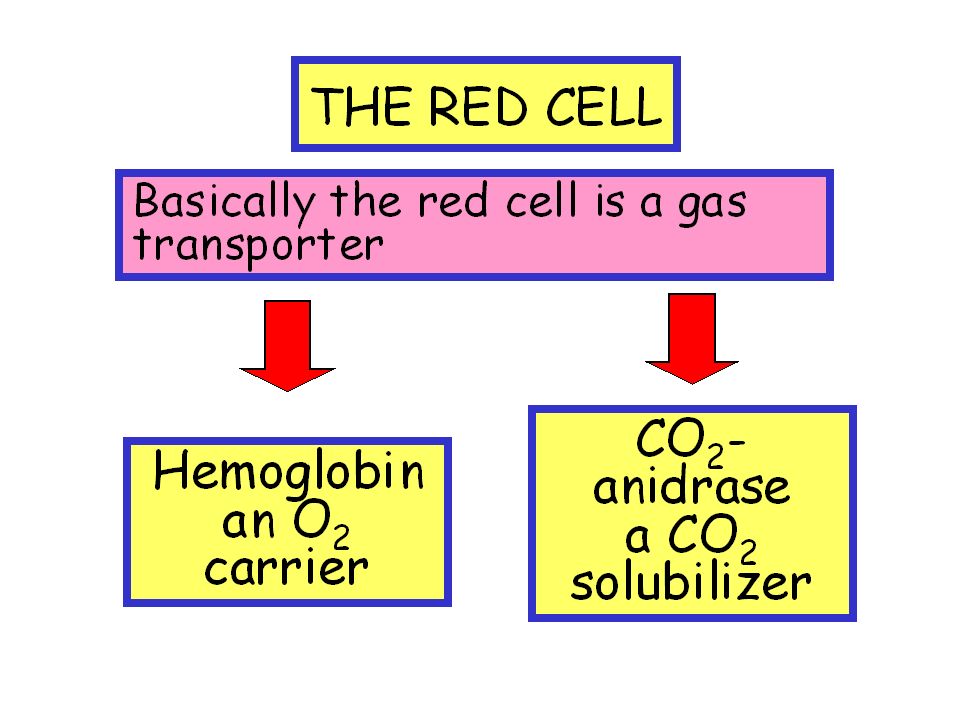
THE RED CELL I The simplest cell in the body (no nucleus, no mitochondria, no RNA) Very few kind of proteins (no HLA, no oxydative metabolism) Very modest interactivity Very modest immunogenicity (no HLA class I)
 Very few kind of proteins (no HLA, no oxydative metabolism) Very modest interactivity Very modest immunogenicity (no HLA class I)")
4
TISSUE O2O2 O2O2 O2O2 HCO 3 - + H + CO 2 O2O2 Arterial blood More Oxyemoglobin less H +, Higher Ph LUNG O2O2 O2O2 O2O2 HCO 3 - + H + CO 2 O2O2 Venous blood Less Oxyemoglobin more H + Lower Ph

6
CAUSES OF ANEMIA IMPAIRED PRODUCTION ACUTE BLOOD LOSS INCREASED DESTRUCTION Stem cell is damaged (not a true RBC disorder) Stem cell is OK but some problems occur during differentiation external internal Of healthy blood cells (due to an external problem) Of a sick RBC Hyporegenerative anemia Hemolytic anemia
 Stem cell is OK but some problems occur during differentiation external internal Of healthy blood cells (due to an external problem) Of a sick RBC Hyporegenerative anemia Hemolytic anemia")
7
HEMOLYTIC ANEMIAS OF HEALTHY BLOOD CELLS Estrinsic causes (usually acquired) Mechanical cause Immunologically mediated Induced by parasites Induced by toxins Mechanical valves/ microcirculation disorders Autoimmune hemolytic anemia/ transfusion reaction Endotoxins/ snake venom malaria
 Mechanical cause Immunologically mediated Induced by parasites Induced by toxins Mechanical valves/ microcirculation disorders Autoimmune hemolytic anemia/ transfusion reaction Endotoxins/ snake venom malaria")
8
HEMOLYTIC ANEMIAS OF SICK BLOOD CELLS Intrinsic causes (usually inherited) Qualitative Hemoglobin defect RBC metabolism defect Quantitative Hemoglobin Defect Cytoskeleton/ membrane defect GAPDH Deficiency Spherocytosis Thalassemia (esp alpha) Hemoglobinopathies
 Qualitative Hemoglobin defect RBC metabolism defect Quantitative Hemoglobin Defect Cytoskeleton/ membrane defect GAPDH Deficiency Spherocytosis Thalassemia (esp alpha) Hemoglobinopathies")
9
CONSEQUENCES OF ANEMIA LACK OF O 2 IN TISSUES AND COMPENSATION EFFORTS BY THE ORGANISM INCREASED PRODUCTION/EXCRET ION OF-RBC ASSOCIATED CATABOLYTES ACUTE HEMOLYTIC SYNDROME IN ANY ANEMIA IN CASE OF INEFFECTIVE ERYTROPOIESIS OR HEMOLYSIS IN CASE OF ACUTE INTRA- VASCULAR HEMOLYSIS

10
RED CELL STRUCTURE Cell membrane (external interactions and transport) Cytoskeleton (structure) Metabolic enzymes (energy and detoxyfication) GAS-transporting system ANY OF THESE STRUCTURES CAN BE DAMAGED BY SPECIFIC DISORDERS: (IMMUNE HEMOLYTIC ANEMIA SPHEROCYTOSIS, GAPDH DEFICIENCY, HEMOGLOBINOPATHIES)
 Cytoskeleton (structure) Metabolic enzymes (energy and detoxyfication) GAS-transporting system ANY OF THESE STRUCTURES CAN BE DAMAGED BY SPECIFIC DISORDERS: (IMMUNE HEMOLYTIC ANEMIA SPHEROCYTOSIS, GAPDH DEFICIENCY, HEMOGLOBINOPATHIES)")
11
Dissociazione ossigeno-emoglobina: normale curva sigmoide che correla la saturazione di emoglobina alla parziale pressione di ossigeno (PaO2) a cui è esposta. La curva è spostata a sinistra (è rilasciato meno ossigeno a ogni data PaO2) da un decremento del 2,3 DPG, da un aumento del pH (effetto Bohr) e se l'HbA è sostituita dall'HbF o da un'emoglobina a più alta affinità. La curva è spostata a destra da un aumento del 2,3 DPG attaccato alla Hb, da un decremento del pH e se l'HbA è sostituita dalla HbS oda una HbM (in cui il ferro emico è stabilizzato nella forma ferrica) o se l'Hb è ossidata a metaemoglobina.
 da un decremento del 2,3 DPG, da un aumento del pH (effetto Bohr) e se l HbA è sostituita dall HbF o da un emoglobina a più alta affinità. La curva è spostata a destra da un aumento del 2,3 DPG attaccato alla Hb, da un decremento del pH e se l HbA è sostituita dalla HbS oda una HbM (in cui il ferro emico è stabilizzato nella forma ferrica) o se l Hb è ossidata a metaemoglobina..")
12
HEMOGLOBIN FUNCTION THE O 2 DISSOCIATION CURVE

13
HEMOGLOBIN FUNCTION THE O 2 DISSOCIATION CURVE Myoglobin Hb monomer Low 2,3DPG High 2,3DPG Low Ph High Ph High pCO 2 Low pCO 2 HbF Presence of CO

15
Production of Hb chains

17
Struttura schematica dellHb delladulto (HbA)
")
18
HEMOGLOBIN STRUCTURE

19
HEMOGLOBIN DISORDERS GAS-TRANSPORTING SYSTEM DISORDERS HEME DISORDERS (porphyrias, sideropenic anemia CO-intoxication) GLOBIN DISORDERS (Thalassemia, structural hemoglobinopathies)
 GLOBIN DISORDERS (Thalassemia, structural hemoglobinopathies)")
20
GLOBIN-DISORDERS QUANTITATIVE DISORDERS (thalassemia) QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY
 QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY")
21
HB DISEASES CAN CAUSE MANY PROBLEMS qualitatively impair the gas trasporting system quantitatively impair the gas transporting system Induce erythroblast or RBC toxicity reduce RBC circulatory performance Sickle-cell disease Hb with altered Hb affinity Thalassemia

22
GAS-TRANSPORTING SYSTEM DISORDERS HEMOGLOBIN DISORDERS Structural Hb disorders Diseases related to an impaired Hb synthesis Structural Hb disorders associated to reduced Hb synthesis an Acquired Hb disorders Persistence of HbF Sickle-cell disease thalassemiaHb Lepore CO- intoxication

23
HEMOGLOBIN DISORDERS GAS-TRANSPORTING SYSTEM DISORDERS HEME DISORDERS (porphyrias, sideropenic anemia CO-intoxication) GLOBIN DISORDERS (Thalassemia, structural hemoglobinopathies)
 GLOBIN DISORDERS (Thalassemia, structural hemoglobinopathies)")
24
GLOBIN-DISORDERS QUANTITATIVE DISORDERS (thalassemia) QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY
 QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY")
25
In some areas there is a clear survival avantage for the preservation of these genes. This is usually associated to a survival advantage for heterozygotes

26
Inherited Hb defects are the most common mendelian genetic disorders: 200,000,000 people for thalassemia CLINICAL SYMPTOMS RANGE FROM SILENT TO LETHAL IN UTERO INCIDENCE Disease severity expected observed SOCIAL RELEVANCE Disease severity

27
The human-anopheles-plasmodium ecosystem

28
THE HEMOGLOBIN DISEASE BELT

29
Beta thalassemia incidence in Italy

30
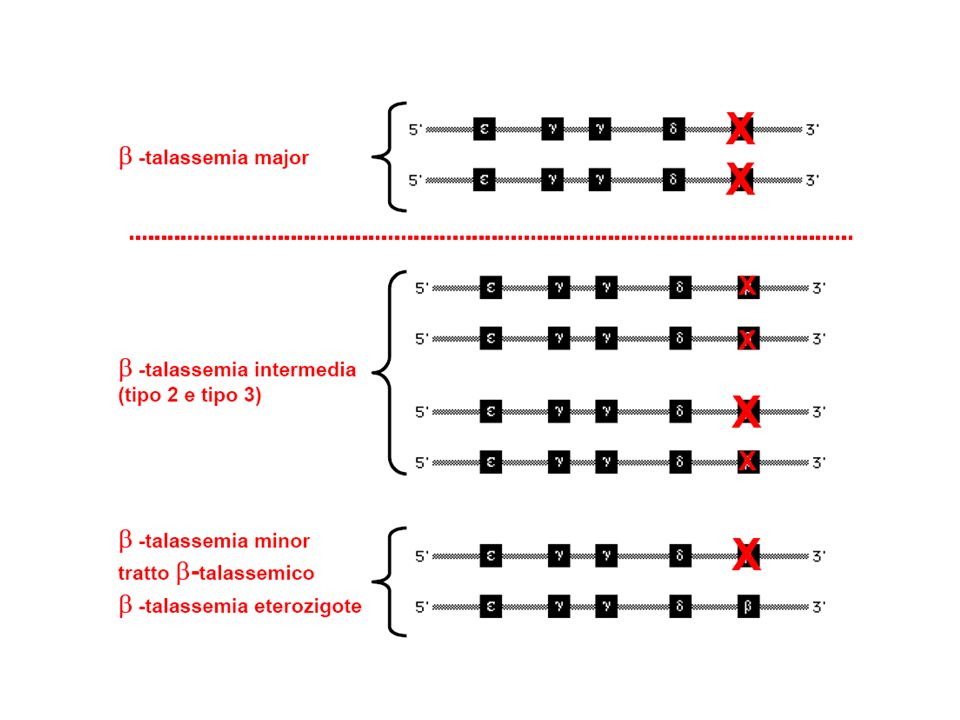
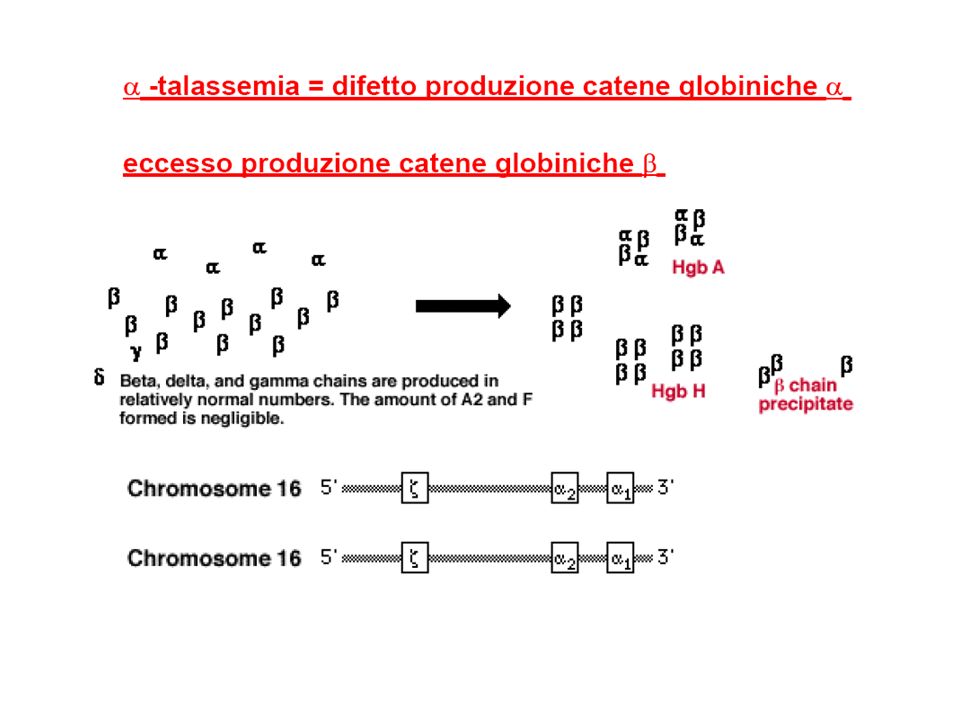
Classificazione dellemoglobinopatie e talassemie: alterazioni strutturali dellHb: anomala polimerizzazione Hb (HbS) anomala cristallizzazione Hb (HbC) emoglobine instabili emoglobine con affinita per lO2 (policitemia); emoglobine con affinita per lO2 (cianosi); difetti quantitativi nella produzione delle catene globiniche -talassemia -talassemia, -talassemia, -talasemia persistenza di Hb fetale (HbF) pancellulare eterocellulare emoglobinopatie acquisite meta-emoglobinemia sulfo-emoglobinemia carbossi-emoglobinemia incremento HbF (chemioterapia, ripresa midollare, mielodisplasie)
 anomala cristallizzazione Hb (HbC) emoglobine instabili emoglobine con affinita per lO2 (policitemia); emoglobine con affinita per lO2 (cianosi); difetti quantitativi nella produzione delle catene globiniche -talassemia -talassemia, -talassemia, -talasemia persistenza di Hb fetale (HbF) pancellulare eterocellulare emoglobinopatie acquisite meta-emoglobinemia sulfo-emoglobinemia carbossi-emoglobinemia incremento HbF (chemioterapia, ripresa midollare, mielodisplasie)")
37
THE HEMOGLOBIN GENES

39
Production of Hb chains

41
DIAGNOSI -TALASSEMIA ETEROZIGOTE - Sospetto per familiarità e/o microcitemia - Lieve anemia (Hb 9,5 – 11,5 g/dl) con GR aumentati (5- 6,5 x 10 6 / l), riduzione MCV (60-75 / l) ed MCH. - Controllare sideremia: se normale eseguire elettroforesi Hb: diagnosi se HbA2 >3-3,5%. - Se sideropenia, normalizzare sideremia prima di elettroforesi.

43
-TALASSEMIA OMOZIGOTE Gravità dell anemia dipende da: - eventuale produzione residua di catene : genotipo 0 / 0 : malattia più grave, produzione solo di HbF e HbA 2 genotipo +/ +: talassemia intermedia - quantità di Hb F e HbA 2 prodotta: malattia molto meno grave se persistenza HbF (es. talassemie con ampie delezioni)
.")
44
TERAPIA -TALASSEMIA OMOZIGOTE TERAPIA DI SUPPORTO - Trasfusioni per mantenere Hb > 10 g/dl: normale crescita e prevenzione deformità scheletriche. - Terapia chelante del ferro con desferrioxamina per infusione s.c. giornaliera di 8- 12 ore. - In fase di sperimentazione chelanti orali. - SOPRAVVIVENZA MEDIANA 35-40 anni. TRAPIANTO DI MIDOLLO ALLOGENICO Se donatore HLA identico e trapianto entro i 10 anni: 80% guarigione definitiva; 10% mortalità.

45
TERAPIA -TALASSEMIA OMOZIGOTE APPROCCI SPERIMENTALI Tentativi di ripristinare la sintesi di HbF mediante riattivazione dei geni con agenti de-metilanti del DNA (5-aza-citidina) e inibitori dell istone – deacetilasi (butirrati e nuove molecole).
 e inibitori dell istone – deacetilasi (butirrati e nuove molecole).")
46
GLOBIN-DISORDERS QUANTITATIVE DISORDERS (thalassemia) QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY
 QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY")
47
HB DEFECTS OFTEN ARE OFTEN ASSOCIATED TO POINT MUTATIONS SOLUBILITY STABILITY Oxygen AFFINITY External AAInternal AAHeme pocket AA

48
The most common qualitative Hb disease affects Hb solubility A PROTOTYPE OF A POINT MUTATION DISEASE 6 Glu--> Val SICKLE CELL DISEASE (HBS) NB 6 Glu-->Lys give rise to HBC with a milder phenotype
 NB 6 Glu-->Lys give rise to HBC with a milder phenotype")
49
THE SAME MUTATION AROSE IN DIFFERENT SUBJECTS AND WAS POSITIVELY SELECTED

50
THE -6 mutation affects Hb sulubility mostly in the deoxygenated state

51
The sickling balance SOLUBLE HB POLYMERIZED HB OXY-state, high PH, good hydration, etc HB F> HB A >HB C > HB S

52
The sickling balance SOLUBLE HB POLYMERIZED HB DEOXY-state, low PH, dehydration, fever, etc HB S > HB C >HB A > HB F

54
SICKLE CELL DISEASE MANIFESTATIONS OCCURRING IN RED CELLS Irreversible sickling and hemolysis MODIFICATION OF MEMBRANE PERMEABILITY MODIFICATION OF ADESIVE PROPERTIES Polimerization of HbS (reversible sickling) INCREASED HB CONCENTRATIONS
 INCREASED HB CONCENTRATIONS")
55
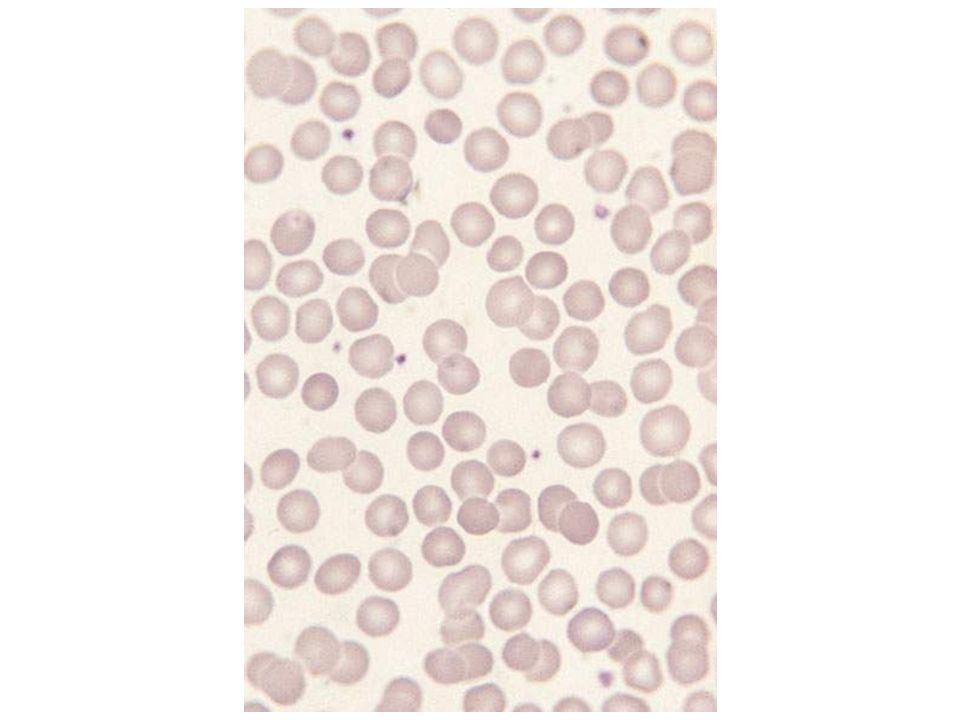
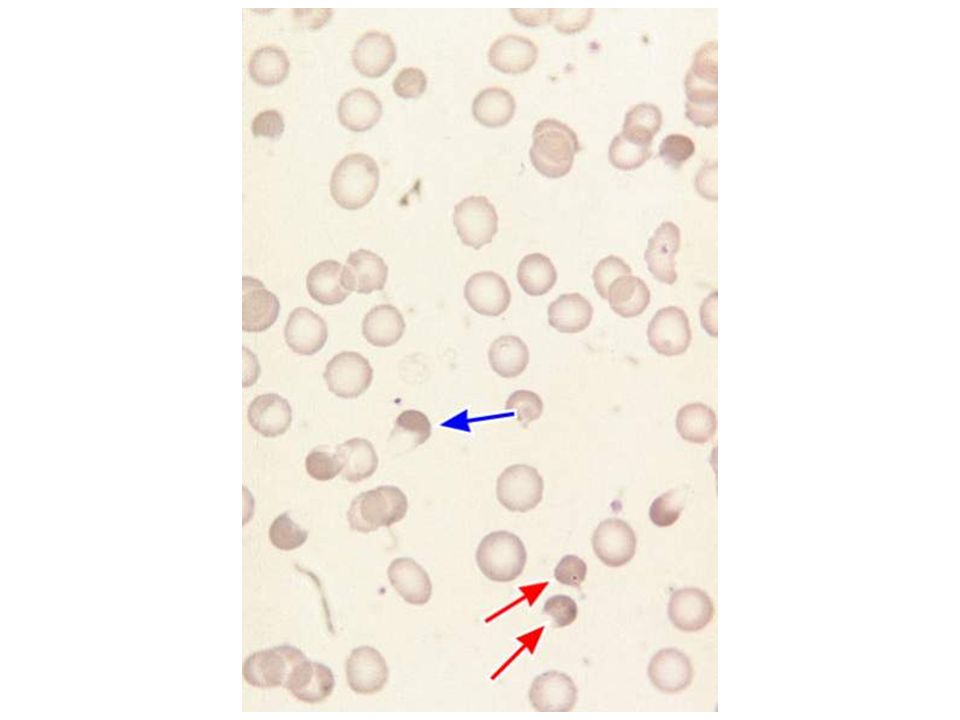
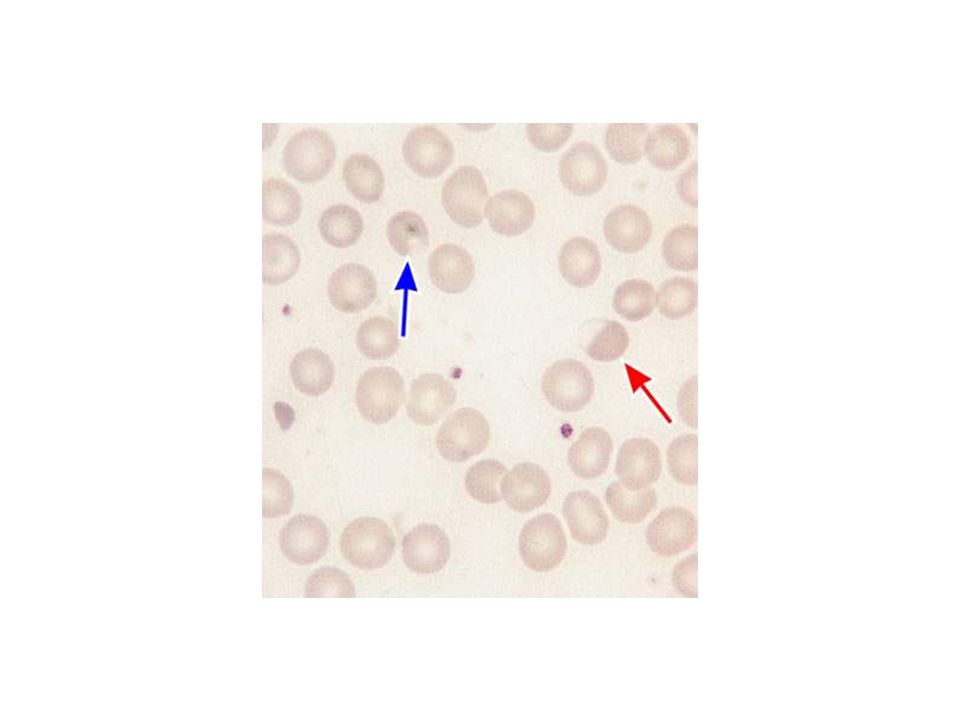
BLOOD SMEAR OF SICKLE CELL DISEASE

56
sickle cell disease: clinical manifestations SICKLING RELATED (pain, ischemia, renal disease etc) HEMOLYTIC (anemia jaundice, gallstones) SPLEEN autoinfarction Aplastic crises SUSCEPTIBILITY TO INFECTIONS
 HEMOLYTIC (anemia jaundice, gallstones) SPLEEN autoinfarction Aplastic crises SUSCEPTIBILITY TO INFECTIONS")
57
sickle cell disease: clinical manifestations Common in black people (8% heterozygotes in african americans) Can be underdiagnosed especially if clinical manifestation are mild It often presents as acute pain syndrome Can be easily diagnosed with a very simple laboratory exam Newborns are usually asymptomatic
 Can be underdiagnosed especially if clinical manifestation are mild It often presents as acute pain syndrome Can be easily diagnosed with a very simple laboratory exam Newborns are usually asymptomatic")
58
QUADRO CLINICO DREPANOCITOSI -Anemia di solito moderata: Hb ~ 8 g/dl, ma con ampie variazioni individuali (5 – 11 g/dl). -QUADRO DOMINATO DAI PROBLEMI VASO- OCCLUSIVI: crisi di intenso dolore osseo o toracico o addominale; infarti ossei con necrosi; ulcere trofiche agli arti inferiori; ischemie ed emorragie cerebrali;

59
QUADRO CLINICO DREPANOCITOSI -infarti polmonari ipertensione polmonare e scompenso cardiaco; -danno renale per ischemia tubulare. -nei primi anni splenomegalia, poi infarti splenici mutipli e fibrosi splenica (autosplenectomia) aumentata suscettibilità ad infezioni batteriche. TUTTE LE SITUAZIONI DI IPOSSIEMIA FANNO PRECIPITARE LE CRISI, ANCHE IN ETEROZIGOTI
 aumentata suscettibilità ad infezioni batteriche. TUTTE LE SITUAZIONI DI IPOSSIEMIA FANNO PRECIPITARE LE CRISI, ANCHE IN ETEROZIGOTI.")
60
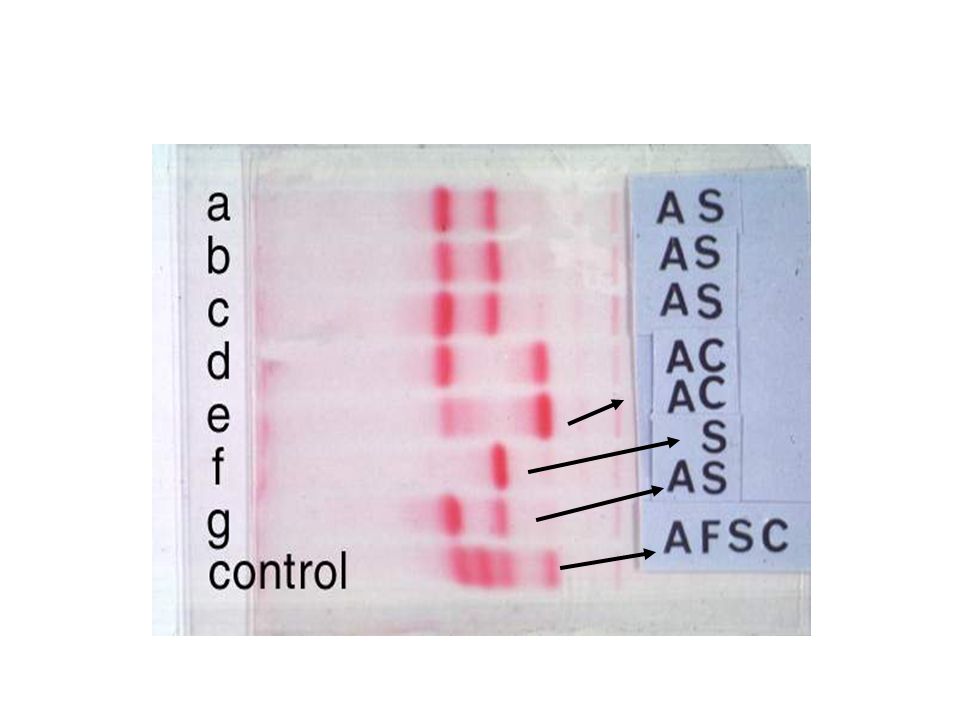
DIAGNOSI DREPANOCITOSI -Sospetto per quadro clinico -Quadro ematologico di anemia emolitica normocitica: reticolociti aumentati, aptoglobina molto ridotta, possibile iperbilirubinemia, frequente riscontro di qualche GR a falce sullo striscio di sangue. - DIAGNOSI CON ELETTROFORESI Hb

62
PROGNOSI DREPANOCITOSI - Un tempo alta mortalità infantile e sopravvivenza massima 30-35 anni -Ora maggior parte pazienti sopravvive fino a 50 anni. -Peggiore negli omozigoti. -Possibile ulteriore miglioramento con nuove terapie

63
TERAPIA DREPANOCITOSI Terapia tradizionale Terapia di supporto: trattamento infezioni, trasfusioni se anemia grave o frequenti episodi ischemici, analgesici oppiacei e idratazione per crisi dolorose, prevenzione ipossiemia. Nuovi approcci -Farmaci miranti a ripristinare HbF: idrossiurea e de-metilanti del DNA (aza-citidina, butirrati): miglioramento quadro clinico. -Trapianto di midollo allogenico: 80-90% di successo ma mortalità intorno 10%
: miglioramento quadro clinico. -Trapianto di midollo allogenico: 80-90% di successo ma mortalità intorno 10%.")
64
A pathogenetic apprach to sickle cell disease: increasing Hb F concentrations

65
SICKLE-CELL COMBINED WITH OTHER HB DISORDERS

66
COMBINED HB DEFECTS HBS/HBE HBS/ tal HBS/HBS + FHB persistence Mild sickle cell disease Moderate to severe sickle cell disease + microcytosis Mild to moderate sickle cell disease HBS/HBA Asymptomatic with exceptions

67
GLOBIN-DISORDERS QUANTITATIVE DISORDERS (thalassemia) QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY
 QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY")
68
GLOBIN-DISORDERS QUANTITATIVE DISORDERS (thalassemia) QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY
 QUALITATIVE DISORDERS (structural hemoglobinopathies) -CHAIN SOLUBILITYSTABILITY Oxygen AFFINITY")
69
HB DEFECTS OFTEN ARE OFTEN ASSOCIATED TO POINT MUTATIONS SOLUBILITY STABILITY Oxygen AFFINITY External AAInternal AAHeme pocket AA or alpha/beta chain interactions

70
A PROTOTYPE OF A POINT MUTATION DISEASE 6 Glu--> Val NB 6 Glu-->Lys give rise to HBC with a milder phenotype (It is better a basic but hydrophilic aa than an hydrophobic one)
")
71
Other HBs with reduced solubility HBD ( 26 Glu--> Gln) very mild HBE ( 26 Glu--> Lys) slightly mild mixed phenotype
 very mild HBE ( 26 Glu--> Lys) slightly mild mixed phenotype")
72
UNSTABLE HEMOGLOBINS Often lethal for the embryo if homozygous, clinically evident in heterozygous (alpha 80%) Hb precipitates (Heinz bodies) with membrane damage and hemolysis (<MCHC) May worsen if particular drugs are given Diagnosis is often difficult with Hb electrophoresis (stability tests) Involves critical internal portions of the Hb chain
 Hb precipitates (Heinz bodies) with membrane damage and hemolysis (<MCHC) May worsen if particular drugs are given Diagnosis is often difficult with Hb electrophoresis (stability tests) Involves critical internal portions of the Hb chain")
73
HBs with altered 0 2 affinity INCREASED AFFINITY REDUCED AFFINITY ERYTROCYTOSISCYANOSIS PM targeting the hb pocket, the 2,3 pg binding site

74
HEMOGLOBIN FUNCTION THE O 2 DISSOCIATION CURVE PO2:100 PO2:50 REDUCED AFFINITY INCREASEDA FFINITY METHEMOGLOBINEMIA

75
HB with decreased oxygen affinity Often lethal for the embryo if clinically evident in heterozygous May be associated with anemia or erythrocytosis Typical feature is cyanosis Dioagnosis is often difficult with Hb electrophoresis (affinity tests) Methemoglobinemia involves two critical His portions of the Hb chain -->Tyr
 Methemoglobinemia involves two critical His portions of the Hb chain -->Tyr")
76
ANEMIA OR ERYTHROCITOSIS RELATE TO THE O 2 DISSOCIATION CURVE PO2:100 PO2:50 ANEMIAERYTHROCYTOSIS

77
HB with increased oxygen affinity Only heterozygous are known Very mild conditions with erythrocytosis Often asymptomatic with only ruddy complexion Dioagnosis is often difficult with Hb electrophoresis (affinity tests) Often the alpha/beta interaction sites are involved
 Often the alpha/beta interaction sites are involved")
78
-THALASSEMIA AND HBF PERSISTENCE WHAT SHOULD BE KNOWN The deletion is bigger in HBF persistence, probably encompassing the region responsible for switchin off the -genes at six months) Relationship between HBF and other hemoglobins (es HBS) A large deletion of the -chain locus is the cause of both disease, one benign and one very severe
 Relationship between HBF and other hemoglobins (es HBS) A large deletion of the -chain locus is the cause of both disease, one benign and one very severe")
79
RED CELL CYTOSKELETON

80
STRUTTURA MEMBRANA ERITROCITARIA

81
CYTOSKELETON DISORDERS VERTICAL DISORDERS (spherocytosis) HORIZONTAL DISORDERS (elliptocytocitosis) LOSS OF CONTACT BETWEEN MEMBRANE AND CYTOSKELETON PERTURBATION IN THE CYTOSKELETAL STRUCTURE WITH IMPAIRED RECOVERY
 HORIZONTAL DISORDERS (elliptocytocitosis) LOSS OF CONTACT BETWEEN MEMBRANE AND CYTOSKELETON PERTURBATION IN THE CYTOSKELETAL STRUCTURE WITH IMPAIRED RECOVERY")
82
DIFETTI MEMBRANA ERITROCITA - Deficit di proteine del citoscheletro sottostante la membrana: anchirina, spectrina, proteina 4.1, proteina 4.2 o della membrana stessa: banda 3. - Sferocitosi - Ellissocitosi - Ovalocitosi - Stomatocitosi

83
Sferocitosi ereditaria, ellissocitosi ereditaria, ovalocitosi Sferocitosi ereditaria Ellissocitosi ereditaria Deficit di anchirina (>50%) Sostituzione aminoacidica Cambiamento di struttura e mutazioni nonsenso Difetti di splicing Delezioni geniche Traslocazioni bilanciate Deficit di spectrina Difetti della catena a (raro) Difetti della catena b (non comune) Anormalità (5%) della pallidina (proteina 4.2) Deficit della banda 3 (20%) Anormalità di spectrina Difetti della catena a (80%) Difetti della catena b (5%) Deficit di proteina 4.1 (15%) Ovalocitosi del Sud-Est dellAsia Difetto della banda 3 (delezione di 9 aminoacidi alla giunzione dei domini citoplasmatico e transmembrana)
 Sostituzione aminoacidica Cambiamento di struttura e mutazioni nonsenso Difetti di splicing Delezioni geniche Traslocazioni bilanciate Deficit di spectrina Difetti della catena a (raro) Difetti della catena b (non comune) Anormalità (5%) della pallidina (proteina 4.2) Deficit della banda 3 (20%) Anormalità di spectrina Difetti della catena a (80%) Difetti della catena b (5%) Deficit di proteina 4.1 (15%) Ovalocitosi del Sud-Est dellAsia Difetto della banda 3 (delezione di 9 aminoacidi alla giunzione dei domini citoplasmatico e transmembrana)")
84
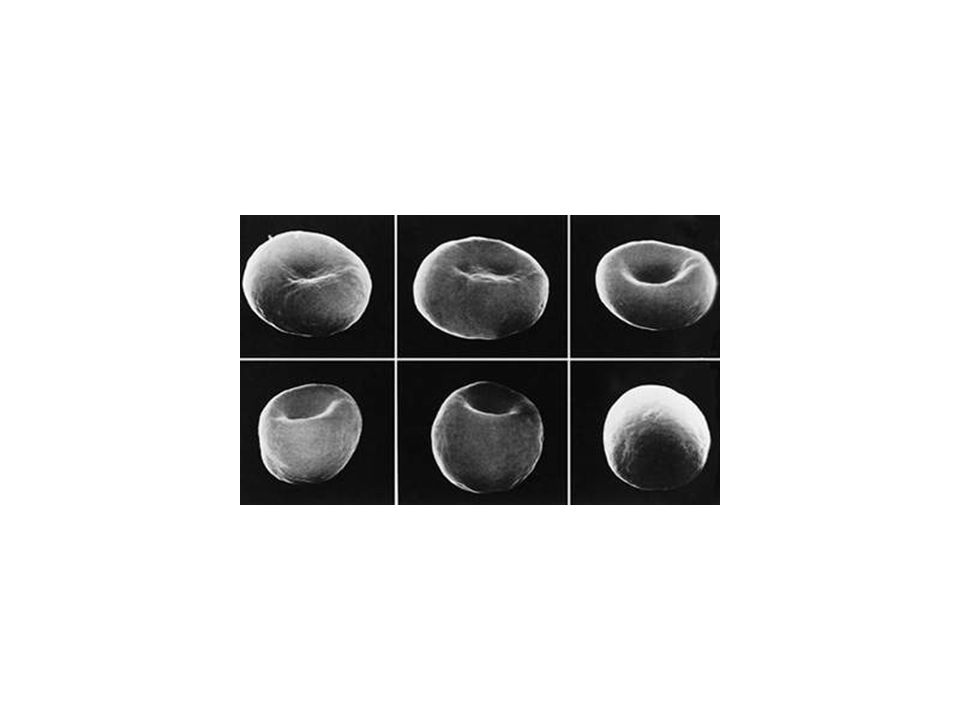
SPHEROCYTOSIS

85
SPHERCYTOSIS (SP) Many vertical defects induce SP. The most common are dominant It is usually characterized by a well compensated hemolytic process with occasional crises MCV is reduced, MCHC is incresed It is basically a disorder associated to membrane losses It is characterized by reduced osmotic resistence and increased autohemolysis It is basically cured with splenectomy

86
SFEROCITOSI EREDITARIA - Più frequente difetto di membrana - Autosomica dominante nel 75% dei casi - Nel 25% casi autosomica recessiva o dominante a penetranza incompleta o neomutazione - Deficit anchirina o spectrina o banda 3 o proteina 4.2

87
SFEROCITOSI EREDITARIA - Deficit di proteine determina perdita di coesione citoscheletro con strato lipidico soprastante - Perdita lipidi, riduzione superficie e assunzione di forma sferica - Eritrociti sferici trattenuti nei capillari splenici. - Emolisi cronica. - Sopravvivenza eritrocitaria variamente ridotta.

90
SFEROCITOSI EREDITARIA QUADRO CLINICO – EMATOLOGICO - Anemia di gravità variabile: da casi asintomatici (Hb ~ 11 g/dl) a casi gravi con dipendenza trasfusionale. -Reticolocitosi spiccata. - Segni di emolisi: iperbilirubinemia indiretta, LDH e urobilinogeno, aptoglobina. - Splenomegalia e calcolosi biliare. - Raramente crisi aplastiche: per deficit folati o infezione da parvovirus blocco temporaneo dell eritropoiesi con grave anemizzazione.

91
SFEROCITOSI EREDITARIA DIAGNOSI - Segni di anemia emolitica iperrigenerativa normo-microcitica. - Morfologia eritrocitaria. - Ipersensibilità eritrocitaria alla lisi osmotica. - Analisi proteine di membrana

92
OSMOTIC RESISTANCE IN SPHEROCYTES AND NORMOCYTES NaCl Concentrations NORMAL % HEMOLYSIS SP Plasma

93
-

94
SFEROCITOSI EREDITARIA TERAPIA SPLENECTOMIA: migliora nettamente la sopravvivenza eritrocitaria. Praticata dopo i 20 anni se anemia moderata (aumenta rischio di sepsi da meningococco, pneumococco, hemophilus, soprattutto in età infantile). Splenectomia nell infanzia solo se anemia grave, previa vaccinazione contro i suddetti batteri.
. Splenectomia nell infanzia solo se anemia grave, previa vaccinazione contro i suddetti batteri..")
96
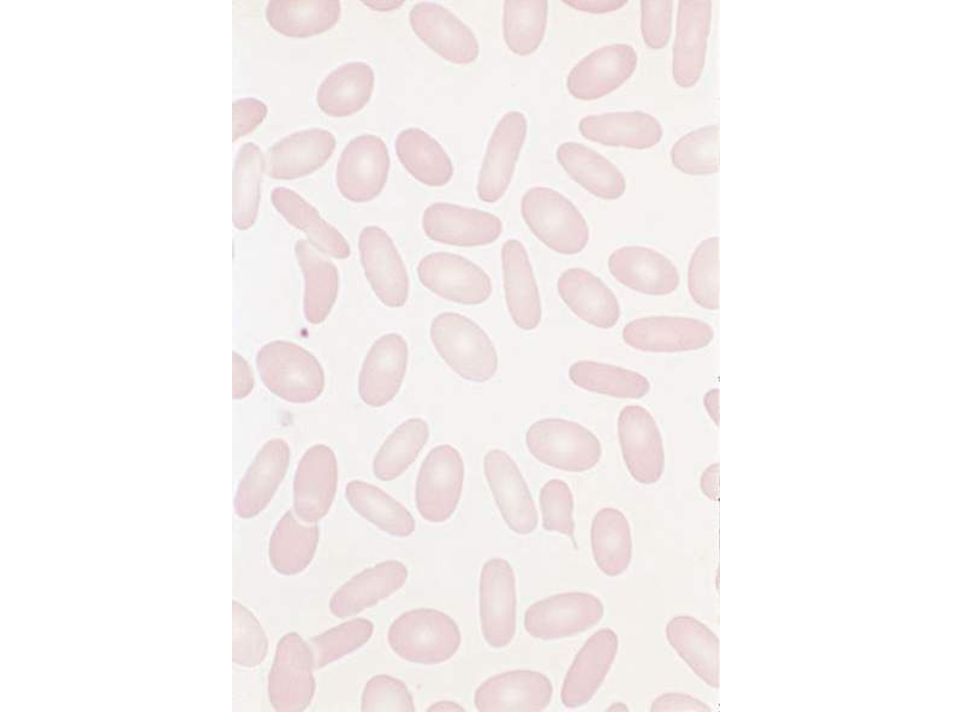
ELLYPTOCYTOSIS

97
ELLIPYOCYTOSIS (EP) Many horizontal defects induce EP. The most common are dominant It is characterized by a very well compensated hemolytic process MCV is not reduced, MCHC is not incresed It is basically a disorder associated to unability to recover from capillary-induced shape modifications NO reduced osmotic resistence Often requires no therapy. Splenectomy is effective if necessary

98
STOMATOCYTOSIS (ST) DISEASE ASSOCIATED TO A PERMEABILITY DEFECT (protein 7.2) IT IS CHARACTERIZED BY A VERY WELL COMPENSATED HEMOLYTIC PROCESS RBC BECOME FATTY. WITH REDUCED MCHC AND INCREASED MCV THERE IS REDUCED OSMOTIC RESISTENCE OFTEN REQUIRES NO THERAPY.

99
METABOLIC DISEASES OF THE RBC: WHAT SHOULD BE KNOWN Know the basic functions of ATP, NADPH and 2,3PG in RBC Describe epidemiology, pathogenesis and clinicakl features of GAPDH and PK deficiencies Explain why many metabolic defects express mostly with anemia Describe briefly the enzymatic pathways of RBC

100
ANEMIE EMOLITICHE PER DIFETTI ENZIMATICI VIE ENZIMATICHE ERITROCITARIE - Glicolisi anerobia: per produzione di ATP - Shunt dei pentosi (esosomonofosfati): per riconvertire glutatione ossidato (GSSG) a glutatione ridotto (GSH) utilizzato per proteggere da ossidazione i gruppi SH dell emoglobina e ridurre Fe +++ (metaemoglobina) a Fe ++
: per riconvertire glutatione ossidato (GSSG) a glutatione ridotto (GSH) utilizzato per proteggere da ossidazione i gruppi SH dell emoglobina e ridurre Fe +++ (metaemoglobina) a Fe ++")
102
ANEMIE EMOLITICHE PER DIFETTI ENZIMATICI - Descritti difetti congeniti per quasi tutte le tappe della glicolisi anaerobia e della produzione di GSH (molti rarissimi): determinano ridotta sopravvivenza eritrociti di varia gravità - Difetti più frequenti a carico di glucosio-6 fosfato deidrogenasi (G6PD) e piruvato chinasi (PK)
: determinano ridotta sopravvivenza eritrociti di varia gravità - Difetti più frequenti a carico di glucosio-6 fosfato deidrogenasi (G6PD) e piruvato chinasi (PK)")
103
DEFICIT G6PD - Due varianti normali principali: G6PD tipo B, più comune, G6PD tipo A, più frequente nelle popolazioni africane. - Molte altre varianti più rare, con attività normale o ridotta. - Varianti mutate più frequenti: G6PD A- e tipo mediterraneo codificano per enzima meno efficiente e ridotto quantitativamente. - Deficit enzimatico più grave nel tipo mediterraneo.

104
DEFICIT G6PD: GENETICA Gene per G6PD su cromosoma X Malattia recessiva, legata al sesso: - maschi emizigoti: sani o malati - femmine omozigoti (sane o malate) o eterozigoti (portatrici)
 o eterozigoti (portatrici)")
105
DEFICIT G6PD: PATOGENESI ANEMIA La scarsa attività enzimatica riduce la disponibilità di NADPH, indispensabile a glutatione-reduttasi per rigenerare GSH da GSSG. In carenza di GSH, Hb suscettibile a stress ossidativi (farmaci): ossidazione dei gruppi SH di cisteina, distacco di EME e precipitazione di globina corpi di Heinz.
: ossidazione dei gruppi SH di cisteina, distacco di EME e precipitazione di globina corpi di Heinz..")
106
DEFICIT G6PD: PATOGENESI ANEMIA - I corpi di Heinz rendono rigido il GR che resta intrappolato nei capillari splenici ed emolizza. - Alcuni GR perdono corpo di Heinz e parte di membrana e tornano in circolo. - Nei casi più gravi emolisi intravascolare anche extra-splenica. - I GR più giovani hanno > corredo enzimatico e sono più resistenti all emolisi.

109
DEFICIT G6PD: QUADRO CLINICO - Quadro di anemia emolitica normocitica ad insorgenza acuta, correlata all esposizione ad alcuni farmaci o alle fave (solo alcuni soggetti con G6PD tipo mediterraneo): Rapida insorgenza di sintomi di anemia, dolori lombari, subittero, urine ipercromiche per alcuni giorni. Nel favismo possibile febbre, emoglobinuria e insufficienza renale acuta. In rari casi, con deficit molto grave: anemia emolitica cronica.

110
DEFICIT G6PD: DIAGNOSI - Esami di laboratorio indicativi di anemia emolitica con reticolocitosi. - Dosaggio attività enzimatica: valore molto ridotto lontano dalla crisi emolitica; durante la crisi possibile valore normale dovuto a GR giovani sopravvissuti.

111
DEFICIT G6PD: TERAPIA PROFILASSI: evitare esposizione a farmaci ossidanti e fave. TERAPIA CRISI EMOLITICA: idratazione, diuretici, trasfusioni se anemia molto grave

112
Deficit di G6PD Farmaci che possono causare anemia emolitica in soggetti con deficit di G6PD Farmaci che possono essere somministrati a soggetti con deficit di G6PD e senza NSHA Antimalarici Pirimetamina con sulfadossina (Fansidar) Pirimetamina con dapsone (Maloprim) Primachina ?Clorochina Sulfonamidi Sulfametossazolo, Altri sulfonamidi Sulfoni Dapsone, Tiazolosulfone Altri composti antibatterici Nitrofurantoina, Acido nalidixico Antielmintici Beta-naftolo Miscellanea ?Vitamina K, Naftalene (palline antitarma) Blu di metilene Doxorubicina, rasburicasi Acido ascorbico Aspirina Colchicina Isoniazide Menadiolo Fenitoina Probenecid Procainamide Pirimetamina Chinidina Chinino Trimetoprima ? - Cè qualche dubbio su questi composti

113
DEFICIT DI PK - Autosomico recessivo. - Produzione di isoenzima qualitativamente inefficiente o quantitativamente ridotto. - Il deficit di piruvato-chinasi determina inefficienza della glicolisi anaerobia e carenza di ATP. - Emolisi cronica per ridotta sopravvivenza eritrocitaria.

114
DEFICIT DI PK - Anemia di gravità variabile, normocromica e normocitica. - Esami di laboratorio indicativi di emolisi. - Test di autoemolisi (incubazione per 48 h a 37°C) positivo. - Attività enzimatica ridotta. -TERAPIA: splenectomia nei casi gravi.
 positivo. - Attività enzimatica ridotta. -TERAPIA: splenectomia nei casi gravi..")


















 is a disease state characterized by airflow limitation that is not fully reversible. The.>")












