Scaricare la presentazione
1
Fisiopatologia della sepsi dall’infiammazione al danno d’organo
Dr. Matteo Giorgi-Pierfranceschi

2
Patogeni nella sepsi Gram negativi Gram positivi Funghi 20% Infezioni miste 45% 19% Infezioni miste 10% 3% 3% Non confermate Solo il 60% dei casi di sepsi severa / shock settico sono associati ad infezioni documentate La progressione della malattia è indipendente dal tipo di patogeno The most common microbial causes of sepsis are pure Gram-negative or Gram- positive pathogens (36% and 35% of cases, respectively).15 Mixed bacterial infections account for 19% of cases, pure fungal 5%, with mixed bacterial/ fungal/viral infections comprising the remaining 5%.15 Almost every type of micro-organism has the potential to initiate an episode of sepsis. However, only 60% of cases may be associated with a microbiologically confirmed infectious pathogen.15 The clinical progression of sepsis is similar, regardless of the infectious agent.6,15,23 Although Gram-negative bacteria have traditionally been the most common bacteria associated with septic shock,25,26 the relative proportion of cases associated with Gram-positive infection has escalated.6,23,24
.15. Mixed bacterial infections account for 19% of cases, pure fungal 5%, with mixed bacterial/ fungal/viral infections comprising the remaining 5%.15. Almost every type of micro-organism has the potential to initiate an episode of sepsis. However, only 60% of cases may be associated with a microbiologically confirmed infectious pathogen.15. The clinical progression of sepsis is similar, regardless of the infectious agent.6,15,23. Although Gram-negative bacteria have traditionally been the most common bacteria associated with septic shock,25,26 the relative proportion of cases associated with Gram-positive infection has escalated.6,23,24.")
3
SEPSI Patogeni nella sepsi INFIAMMAZIONE risposta immune
Gram - Candida alb. Funghi Gram + e.g. Neisseria meningitidis Escherichia coli e.g. Staphylococcus aureus Streptococcus pneumoniae Enterococcus faecalis Ergosteroli Endotossine (LPS) ed altre tossine Componenti della parete cellulare (ac. Lipoteicoico) ed altri componenti risposta immune risposta immune Gram-negative and Gram-positive bacteria initiate the disease process by different mechanisms.6 However, there is an overlap in some inflammatory pathways. The CD14 receptor, present on many human cell types, plays a key role in initiating cellular activation by bacterial envelope components from both Gram-negative and Gram-positive bacteria.6 Despite subtle differences in the host response to Gram-positive and Gram-negative pathogens, disease severity and mortality in sepsis is similar.6,23,24 Despite the apparent differences between Gram-negative and Gram-positive bacteria, the systemic inflammatory responses for both types of pathogen are, in a qualitative sense, similar.6,23 This is important because the identity of the causative organism is often unknown at the onset of sepsis and treatment targeting the host inflammatory response needs to be rapid and beneficial, regardless of the causative organism.6,27 INFIAMMAZIONE SEPSI
 ed altre tossine. Componenti della parete cellulare (ac. Lipoteicoico) ed altri componenti. risposta immune. risposta immune. Gram-negative and Gram-positive bacteria initiate the disease process by different mechanisms.6 However, there is an overlap in some inflammatory pathways. The CD14 receptor, present on many human cell types, plays a key role in initiating cellular activation by bacterial envelope components from both Gram-negative and Gram-positive bacteria.6. Despite subtle differences in the host response to Gram-positive and Gram-negative pathogens, disease severity and mortality in sepsis is similar.6,23,24. Despite the apparent differences between Gram-negative and Gram-positive bacteria, the systemic inflammatory responses for both types of pathogen are, in a qualitative sense, similar.6,23. This is important because the identity of the causative organism is often unknown at the onset of sepsis and treatment targeting the host inflammatory response needs to be rapid and beneficial, regardless of the causative organism.6,27. INFIAMMAZIONE. SEPSI.")
4
Sepsi da Gram-negativi: meccanismi patogenetici
Una endotossina della parete cellulare lipopolisaccaride (LPS) – è il fattore chiave associato alla tossicità dei batteri Gram-negativi LPS è formato da: lipid A core region O-antigen polysaccharide Despite the existence of many virulent factors associated with Gram-negative bacteria, current evidence indicates that endotoxin is the principal component responsible for initiating the process of sepsis.6 Lipopolysaccharide (LPS) is the active component of this endotoxin and the factor most commonly associated with toxicity.28 LPS comprises three distinct structural regions: (a) lipid A – the toxic moiety, (b) the core region – a series of saccharide residues (c) O-antigen polysaccharide.6,28 Many of the immunological effects of LPS are mediated by their activation of macrophages and subsequent release of immunological products.28 Endotoxin (lipopolysaccharide [LPS]), a major constituent of Gram-negative bacterial cell walls, is a complex macromolecule and one of the most important microbial products implicated in sepsis.29 Following infection, LPS binds to serum LPS-binding protein, which increases the clearance of LPS and facilitates its interaction with a cell surface receptor (CD14) expressed by leucocytes (e.g. monocytes, macrophages and neutrophils). Thereafter, LPS activates both the classical and alternative complement pathways.30
 – è il fattore chiave associato alla tossicità dei batteri Gram-negativi. LPS è formato da: lipid A. core region. O-antigen polysaccharide. Despite the existence of many virulent factors associated with Gram-negative bacteria, current evidence indicates that endotoxin is the principal component responsible for initiating the process of sepsis.6. Lipopolysaccharide (LPS) is the active component of this endotoxin and the factor most commonly associated with toxicity.28. LPS comprises three distinct structural regions: (a) lipid A – the toxic moiety, (b) the core region – a series of saccharide residues (c) O-antigen polysaccharide.6,28. Many of the immunological effects of LPS are mediated by their activation of macrophages and subsequent release of immunological products.28. Endotoxin (lipopolysaccharide [LPS]), a major constituent of Gram-negative bacterial cell walls, is a complex macromolecule and one of the most important microbial products implicated in sepsis.29 Following infection, LPS binds to serum LPS-binding protein, which increases the clearance of LPS and facilitates its interaction with a cell surface receptor (CD14) expressed by leucocytes (e.g. monocytes, macrophages and neutrophils). Thereafter, LPS activates both the classical and alternative complement pathways.30.")
5
Sepsi da Gram-positivi: meccanismi patogenetici
Componenti della parete cellulare Lipoteichoic acid Peptidoglycan Prodotti extracellulari A–B toxins (diphtheria) Pore-forming toxins (-toxin of Staphylococcus aureus) Hydrolysing enzymes (proteases, hyaluronidases etc) Superantigens (TSST, SPEA etc) Gram-positive bacteria harbour a range of toxins, employing a diverse range of toxic mechanisms.6,30 Some similarities have been discovered in the immune responses to some Gram- negative and Gram-positive bacterial toxins.31 Lipoteichoic acid from Gram-positive bacteria demonstrates CD14-dependent (receptor present on many human cell types) cellular activation, thus also stimulating leucocytes by a similar mechanism to Gram-negative bacteria.31 Furthermore, Gram-positive bacteria posess an array of other factors that contribute to evasion of the host immune response, such as antiphagocytic capsules, intrinsic resistance to bacteriolysis and invasive properties.6 Some studies in animal models have indicated that the cytokine profile triggered by exotoxins may be different to that initiated in response to endotoxins, though the few studies performed in sepsis patients have been inconclusive.6 TSST: toxic shock syndrome toxin, SPEA: streptococcal pyrogenic extoxin A
 Pore-forming toxins (-toxin of Staphylococcus aureus) Hydrolysing enzymes (proteases, hyaluronidases etc) Superantigens (TSST, SPEA etc) Gram-positive bacteria harbour a range of toxins, employing a diverse range of toxic mechanisms.6,30. Some similarities have been discovered in the immune responses to some Gram- negative and Gram-positive bacterial toxins.31. Lipoteichoic acid from Gram-positive bacteria demonstrates CD14-dependent (receptor present on many human cell types) cellular activation, thus also stimulating leucocytes by a similar mechanism to Gram-negative bacteria.31 Furthermore, Gram-positive bacteria posess an array of other factors that contribute to evasion of the host immune response, such as antiphagocytic capsules, intrinsic resistance to bacteriolysis and invasive properties.6 Some studies in animal models have indicated that the cytokine profile triggered by exotoxins may be different to that initiated in response to endotoxins, though the few studies performed in sepsis patients have been inconclusive.6. TSST: toxic shock syndrome toxin, SPEA: streptococcal pyrogenic extoxin A.")
6
altri patogeni Candida spp. fungo patogeno maggiormente correlato alla sepsi (mortalità 50%) Virus ( RSV, influenza virus) o parassiti (malaria) possono sostenere quadri di sepsi Sepsis caused by systemic Candida infection is associated with a high mortality rate of up to 50% in the USA.39 Candida spp. (e.g. C. albicans, C. glabrata, C. tropicalis, C. parapsilosis) are the most common fungal pathogens associated with sepsis. These organisms are difficult to diagnose and the mortality rate is high.39 Studies have shown that Candidaemia is associated with high mortality. However, larger studies are needed in this area to confirm this finding.40 RSV: respiratory syncytial virus
 o parassiti (malaria) possono sostenere quadri di sepsi. Sepsis caused by systemic Candida infection is associated with a high mortality rate of up to 50% in the USA.39. Candida spp. (e.g. C. albicans, C. glabrata, C. tropicalis, C. parapsilosis) are the most common fungal pathogens associated with sepsis. These organisms are difficult to diagnose and the mortality rate is high.39 Studies have shown that Candidaemia is associated with high mortality. However, larger studies are needed in this area to confirm this finding.40. RSV: respiratory syncytial virus.")
7
Risposta all’infezione Progressione da sepsi a sepsi severa e shock
Patogeno Infezione Risposta dell’ospite altri fattori Coagulaz/ fibrinolisi Infiammaz. disfunzione endoteliale risposta sistemica Perdita della omeostasi Disfunzione d’organo morte Bacterial infection evokes a host response by triggering the immune system. Normally, the endothelium plays a key role in maintaining homeostasis.41,42 Systemic microvascular endothelial damage compromises the active, moderating role of the endothelium.41 In the sepsis patient, endothelial dysfunction is central to the imbalance between inflammation, coagulation and fibrinolysis.41 Other pathophysiological mechanisms, including nitric oxide and mitochondrial dysfunction, may also be involved in disease progression.43 Loss of homeostasis in sepsis patients may result in organ dysfunction.41,44

8
omeostasi Infiammazione Coagulazione Fibrinolisi omeostasi
There are many complex factors and mechanisms involved in maintaining homeostasis.41,44,46,47 Homeostasis represents a fine balance of internal regulation involving many physiological mechnisms, such as inflammation, coagulation and fibrinolysis.41,44,46,47 omeostasi

9
Perdita dell’omeostasi in corso di sepsi
Fibrinolisi Infiammazione Coagulazione Disruption of regulation of these processes, such as in severe sepsis, affects this balance and homeostasis is lost.44,46–48 Severe sepsis is a procoagulant state associated with reduced fibrinolysis.49 Ultimately, this deleterious combination appears to create a dynamic process of coagulopathy.50 There is an uncontrolled, positive feedback loop of inflammation and coagulation. The interplay of these malfunctioning mechanisms is now thought to be a driving force behind progressive organ dysfunction and mortality in severe sepsis.46,47,51 Perdita dell’omeostasi Disfunzione endoteliale Danno multiorgano

10
Ruolo dell’infiammazione nella sepsi
Risposta dell’ospite Patogeno Infezione altri fattori Infiammazion disfunzione endoteliale Coagulazion/ fibrinolisi Perdita omeostasi Disfunz d’organo Morte

11
Inflammatory response to microbial toxins
Pathogen Infection T-cell Endothelial damage Platelet activation – aggregation Neutrophil activation, aggregation, degranulation; release of oxygen free radicals and proteases Anti-inflammatory mediators IL-4, IL-10, IL-11, IL-13, IL-1r Pro-inflammatory cytokines Tumour necrosis factor TNF, IL-1, IL-6, IL-8 IL-2, Interferon-, GM-CSF Monocyte/ macrophage Stimulation of the host immune response by toxins causes an inflammatory response that can lead to endothelial damage.41 Pro-inflammatory cytokines, such as tumour necrosis factor (TNF), interleukin-1 (IL-1) and IL-6, are released in response to infection with the aim of destroying damaged tissue and promoting wound repair. Normally, anti-inflammatory mediators (e.g. IL-10, IL-13) are subsequently released to regulate the inflammatory response and restore homeostasis.48 Although inflammation is an essential host response, excessive levels of pro-inflammatory cytokines can lead to systemic endothelial damage, and high levels of anti-inflammatory mediators can result in immune suppression.48 In sepsis, the imbalance between pro-inflammatory cytokines and anti-inflammatory mediators results in: • stimulated coagulation response52 • inhibited anticoagulant response44,47 • inhibited fibrinolytic response.44,47,53,54 These processes lead to endothelial damage and loss of equilibrium between the coagulation and fibrinolytic mechanisms.44 As a procoagulant state develops, thromboses may form in the microvasculature, and in severe sepsis, the condition may progress to acute organ dysfunction and eventually death.44,47,55 IL: interleukin, GM-CSF: granulocyte macrophage colony-stimulating factor
, interleukin-1 (IL-1) and IL-6, are released in response to infection with the aim of destroying damaged tissue and promoting wound repair. Normally, anti-inflammatory mediators (e.g. IL-10, IL-13) are subsequently released to regulate the inflammatory response and restore homeostasis.48. Although inflammation is an essential host response, excessive levels of pro-inflammatory cytokines can lead to systemic endothelial damage, and high levels of anti-inflammatory mediators can result in immune suppression.48. In sepsis, the imbalance between pro-inflammatory cytokines and anti-inflammatory mediators results in: • stimulated coagulation response52. • inhibited anticoagulant response44,47. • inhibited fibrinolytic response.44,47,53,54. These processes lead to endothelial damage and loss of equilibrium between the coagulation and fibrinolytic mechanisms.44 As a procoagulant state develops, thromboses may form in the microvasculature, and in severe sepsis, the condition may progress to acute organ dysfunction and eventually death.44,47,55. IL: interleukin, GM-CSF: granulocyte macrophage colony-stimulating factor.")
12
Infiammazione: adesione dei leucociti all’endotelio
Tumour necrosis factor (TNF) Cytokines: IL-6, IL-8, IL-10 Oxygen free radicals Platelet activating factor Proteases Prostaglandins Leukotrienes Bradykinin Inflammation Coagulation Suppressed fibrinolysis TNF IL-1 Monocita Danno endoteliale Neutrofilo Endothelium Adesione trombosi microvascolare Tissue factor IL-6, IL-8 Chemotactic mediators released from the site of inflammation attract leucocytes.41,51 Endothelial damage results in the production of tissue factor (TF).51 Plasma levels of endogenous anticoagulants are depleted.51,55 Plasminogen activator inhibitor-1 (PAI-1), a key inhibitor of fibrinolysis, is activated.44,47 Leucocyte adhesion to endothelial cells is a critical step in the inflammatory process. Selectins, stimulated by tumour necrosis factor and interleukin-1, aid this adhesion. Selectins further promote inflammation and coagulation by mediating the interaction of leucocytes and platelets.41,55 Endothelial damage caused by excessive pro-inflammatory cytokine activity results in the release of TF.51 TF is a key mediator between the immune system and coagulation pathways, and leads to the deposition of fibrin.51 Damage to the vascular endothelium leads to suppression of anticoagulant factors and endogenous plasma proteins, such as protein C, heparin sulphate and antithrombin. Thus, the anticoagulant response is suppressed.51,55 Pro-inflammatory cytokines also induce expression of PAI-1, an enzyme that plays a major role in inhibition of fibrinolysis by stopping the production of the plasmin needed for dissolution of blood clots.47 Site of inflammation Chemotactic mediators attract leucocytes to site of inflammation Macrofagi IL: interleukin
 Cytokines: IL-6, IL-8, IL-10. Oxygen free radicals. Platelet activating factor. Proteases. Prostaglandins. Leukotrienes. Bradykinin. Inflammation Coagulation Suppressed fibrinolysis. TNF. IL-1. Monocita. Danno endoteliale. Neutrofilo. Endothelium. Adesione. trombosi microvascolare. Tissue factor. IL-6, IL-8. Chemotactic mediators released from the site of inflammation attract leucocytes.41,51. Endothelial damage results in the production of tissue factor (TF).51. Plasma levels of endogenous anticoagulants are depleted.51,55. Plasminogen activator inhibitor-1 (PAI-1), a key inhibitor of fibrinolysis, is activated.44,47. Leucocyte adhesion to endothelial cells is a critical step in the inflammatory process. Selectins, stimulated by tumour necrosis factor and interleukin-1, aid this adhesion. Selectins further promote inflammation and coagulation by mediating the interaction of leucocytes and platelets.41,55. Endothelial damage caused by excessive pro-inflammatory cytokine activity results in the release of TF.51 TF is a key mediator between the immune system and coagulation pathways, and leads to the deposition of fibrin.51. Damage to the vascular endothelium leads to suppression of anticoagulant factors and endogenous plasma proteins, such as protein C, heparin sulphate and antithrombin. Thus, the anticoagulant response is suppressed.51,55. Pro-inflammatory cytokines also induce expression of PAI-1, an enzyme that plays a major role in inhibition of fibrinolysis by stopping the production of the plasmin needed for dissolution of blood clots.47. Site of inflammation Chemotactic mediators attract leucocytes to site of inflammation. Macrofagi. IL: interleukin.")
13
Perdita del controllo dell’ifiammazione nella sepsi
RISPOSTA INFIAMMATORIA mediatori e citochine anti-infiammatorie/pro-infiammatorie Hypo-responsive Overwhelming sepsis Excess compensation/CARS Immunosuppression Increased susceptibility to secondary infection Balanced response Hyper-responsive Sepsis, severe sepsis and MODS If infection is sufficiently severe, pro-inflammatory cytokine release is followed by anti-inflammatory mediator release. This compensatory anti-inflammatory response syndrome (CARS) normally serves to downregulate the initial pro-inflammatory response.60 In sepsis, the body loses its ability to regulate the balance between pro-inflammatory cytokine and anti-inflammatory mediator release. This loss of immune regulation first leads to excessive inflammation systemic inflammatory response syndrome (SIRS) and then excessive immunosuppression (CARS) in an attempt to restore homeostasis.10 CARS can result in secondary infection, causing further complications and a new cycle of unbalanced immune responses.10 The ability to activate and then eventually downregulate the inflammatory response to infection is a vital immune process and it is this ability that is lost in sepsis and severe sepsis.61 In sepsis, there is an initial excessive systemic response to infection resulting in systemic inflammation and coagulation. After these first pro-inflammatory cytokines are released, the body mounts an anti-inflammatory response in an attempt to regain homeostasis. However, in sepsis, the body loses its regulatory ability and an inappropriate CARS is activated.61 CARS may result in excessive immunosuppression through over-expression of anti-inflammatory mediators, such as interleukin-4 (IL-4) and interleukin-10 (IL-10). In this state, the patient is in danger of secondary infections, which would further complicate the existing underlying infection and start a new cycle of excessive immune responses.61 This continuing ‘out-of-balance’ response of the immunomodulatory system can lead to acute organ dysfunction, eventual organ failure and ultimately death. In patients with this ‘immunologic dissonance’, it is therefore vital to try and regain homeostasis and organ function.10 morte risoluzione morte CARS: compensatory anti-inflammatory response syndrome, MODS: multi-organ dysfunction syndrome
 normally serves to downregulate the initial pro-inflammatory response.60. In sepsis, the body loses its ability to regulate the balance between pro-inflammatory cytokine and anti-inflammatory mediator release. This loss of immune regulation first leads to excessive inflammation systemic inflammatory response syndrome (SIRS) and then excessive immunosuppression (CARS) in an attempt to restore homeostasis.10. CARS can result in secondary infection, causing further complications and a new cycle of unbalanced immune responses.10. The ability to activate and then eventually downregulate the inflammatory response to infection is a vital immune process and it is this ability that is lost in sepsis and severe sepsis.61. In sepsis, there is an initial excessive systemic response to infection resulting in systemic inflammation and coagulation. After these first pro-inflammatory cytokines are released, the body mounts an anti-inflammatory response in an attempt to regain homeostasis. However, in sepsis, the body loses its regulatory ability and an inappropriate CARS is activated.61. CARS may result in excessive immunosuppression through over-expression of anti-inflammatory mediators, such as interleukin-4 (IL-4) and interleukin-10 (IL-10). In this state, the patient is in danger of secondary infections, which would further complicate the existing underlying infection and start a new cycle of excessive immune responses.61. This continuing ‘out-of-balance’ response of the immunomodulatory system can lead to acute organ dysfunction, eventual organ failure and ultimately death. In patients with this ‘immunologic dissonance’, it is therefore vital to try and regain homeostasis and organ function.10. morte. risoluzione. morte. CARS: compensatory anti-inflammatory response syndrome, MODS: multi-organ dysfunction syndrome.")
14
The role of endothelial dysfunction in sepsis
Risposta dell’ospite Patogeno Infezione Inflammation Endothelial dysfunction Coagulation/ fibrinolysis Other factors perdita omeostasi disfunz. organo morte

15
Il ruolo dell’endotelio
Interazione con leucociti Rilascio di citochine e mediatori infiammatori Rilascio di mediatori vasomotori(vasodilatazione e vasocostrizione) Effetti sulla cascata coagulativa Aumentata permeabilità edema interstiziale The endothelium plays a key role in maintaining homeostasis.41,42 Damaged endothelium exacerbates the inflammatory response.41,42 The endothelium is a dynamic participant in cellular and organ function. Through the expression of surface proteins and soluble mediators, the endothelium plays an important role in maintaining the physiological equilibrium between coagulation and fibrinolysis. Derangements in these normal functions contribute to systemic inflammation.41,42 During the inflammatory response, endothelial cell activation causes overall vasodilation, allowing leucocytes to access infection sites.41 Normally, the endothelium would downregulate this initial pro-inflammatory response by expressing anti-inflammatory and anticoagulant mediators.42 In sepsis, this regulatory function of the endothelium fails, leading to excessive vasodilation, hypoperfusion, generalised tissue damage and inappropriate cytokine response. The damaged endothelium loses its homeostatic balance and becomes procoagulant, precipitating coagulopathy and microthrombus formation.41 Tissue injury Formation of fibrin clot
 Effetti sulla cascata coagulativa. Aumentata permeabilità edema interstiziale. The endothelium plays a key role in maintaining homeostasis.41,42. Damaged endothelium exacerbates the inflammatory response.41,42. The endothelium is a dynamic participant in cellular and organ function. Through the expression of surface proteins and soluble mediators, the endothelium plays an important role in maintaining the physiological equilibrium between coagulation and fibrinolysis. Derangements in these normal functions contribute to systemic inflammation.41,42. During the inflammatory response, endothelial cell activation causes overall vasodilation, allowing leucocytes to access infection sites.41 Normally, the endothelium would downregulate this initial pro-inflammatory response by expressing anti-inflammatory and anticoagulant mediators.42. In sepsis, this regulatory function of the endothelium fails, leading to excessive vasodilation, hypoperfusion, generalised tissue damage and inappropriate cytokine response. The damaged endothelium loses its homeostatic balance and becomes procoagulant, precipitating coagulopathy and microthrombus formation.41. Tissue injury. Formation of fibrin clot.")
16
Ruolo della coagulazione e della fibrinolisi nella sepsi
Risposta dell’ospite Patogeno Infezione Other factors Inflammation Endothelial dysfunction Coagulation/ fibrinolysis perdita omeostasi disfunz. d’organo morte

18
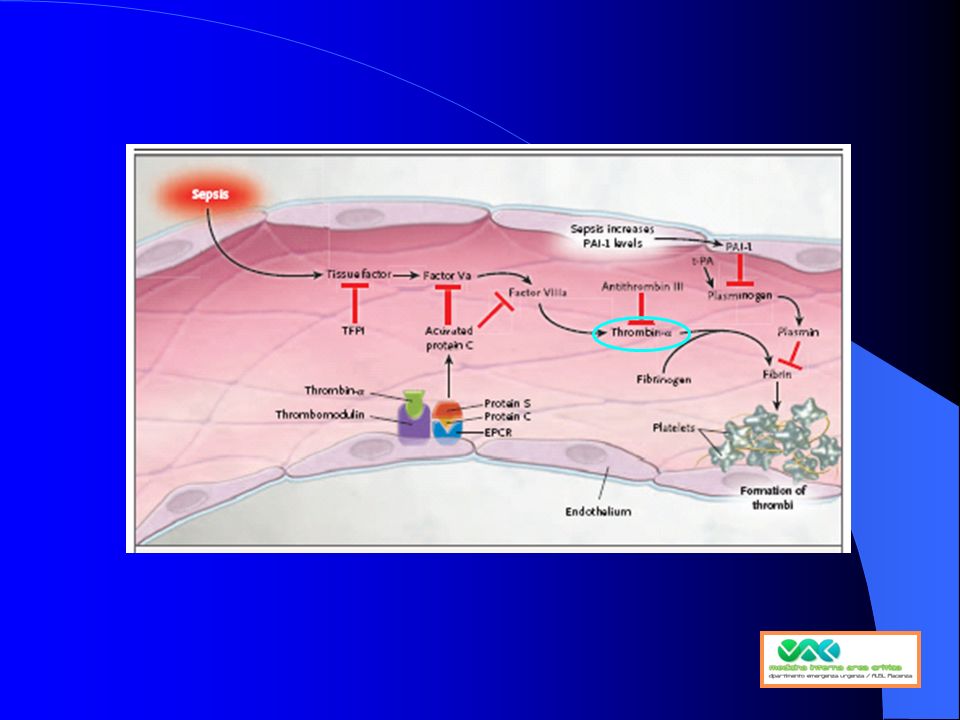
ruolo centrale della trombina nella patogenesi della sepsi
trombosi microvascolare stato procoagulante Infiammaz. Aggregaz / adesione leucociti Formazione clot fibrina attivazione piastrinica Ridotta fibrinolisi Trombina Coagulazione: via comune Tissue factor disfunzione endoteliale Thrombin is a protein with a central role in coagulation.55,62,63 Sepsis patients show clear signs of coagulation activation.51 Markedly increased clotting in the microvasculature has been suggested in sepsis patients.64 Thrombin also promotes further inflammation by increasing platelet activation, platelet aggregation and neutrophil and monocyte adhesion to the endothelium.55,63 Blood coagulation involves a total of 12 coagulation factors that are synthesised in the liver and released into the blood. Some are released by platelets and one (tissue factor) is released from damaged tissue cells.65 The pivotal coagulation factor is thrombin.62 Once thrombin formation is initiated, a cascade of reactions follows and a positive thrombin feedback loop develops resulting in further thrombin formation. Thrombin is the driving force behind the coagulation cascade as it converts fibrinogen to insoluble fibrin threads, increases platelet aggregation and forms stable, tight blood clots.65 Patients with severe sepsis show clear signs of coagulation activation:51,64 levels of fibrin breakdown products are increased, therefore indicating active blood clotting. These patients also appear to have a reduced antithrombotic response (reduced levels of antithrombin and endogenous activated protein C) in order to balance this procoagulant state. This resulting procoagulant state may lead to an inability to clear thromboses in the microvasculature.49,62,64
 is released from damaged tissue cells.65. The pivotal coagulation factor is thrombin.62 Once thrombin formation is initiated, a cascade of reactions follows and a positive thrombin feedback loop develops resulting in further thrombin formation. Thrombin is the driving force behind the coagulation cascade as it converts fibrinogen to insoluble fibrin threads, increases platelet aggregation and forms stable, tight blood clots.65. Patients with severe sepsis show clear signs of coagulation activation:51,64 levels of fibrin breakdown products are increased, therefore indicating active blood clotting. These patients also appear to have a reduced antithrombotic response (reduced levels of antithrombin and endogenous activated protein C) in order to balance this procoagulant state. This resulting procoagulant state may lead to an inability to clear thromboses in the microvasculature.49,62,64.")
19
Ridotta fibrinolisi nella sepsi TAFI e PAI-1
Trombina Pro-coagulant mechanism TAFI fibrinolisi ridotta PAI-1 Persistenza Microtrombi In severe sepsis, fibrinolysis is impaired, thus exacerbating coagulopathy.44,54 Reduced fibrinolysis results in the inability to remove formed thrombi.44 Levels of plasminogen activation inhibitor-1 (PAI-1), a key biomarker of fibrinolysis, are increased in sepsis patients.47,51 Thrombin formation causes an increase in production of thrombin activatable fibrinolysis inhibitor (TAFI), which suppresses fibrinolysis.63 Suppression of fibrinolysis, as shown by raised PAI-1 levels, correlates with a greater risk of mortality.64 In severe sepsis, coagulopathy is exacerbated by suppression of fibrinolysis.54 The activated procoagulant mechanism produces thrombin activatable fibrinolysis inhibitor (TAFI), which suppresses fibrinolysis.63 Tissue factor (TF), released into the plasma as a consequence of endothelial damage, activates PAI-1, which inhibits the formation of plasmin.62 Microthrombi cannot be removed; microvasculature and tissue perfusion is diminished and may cause ischaemia and hypoxia.62 The disruption to fibrinolysis is demonstrated by increased levels of the biomarker PAI-1.51 There is also evidence to suggest that the more marked the coagulation activation and fibrinolysis suppression, the greater the risk of mortality.51,64 TNF-, IL-1 disfunzione endoteliale TAFI: tissue activatable fibrinolysis inhibitor, PAI-1: plasminogen activation inhibitor-1, TNF: tumour necrosis factor, IL: interleukin
, a key biomarker of fibrinolysis, are increased in sepsis patients.47,51. Thrombin formation causes an increase in production of thrombin activatable fibrinolysis inhibitor (TAFI), which suppresses fibrinolysis.63. Suppression of fibrinolysis, as shown by raised PAI-1 levels, correlates with a greater risk of mortality.64. In severe sepsis, coagulopathy is exacerbated by suppression of fibrinolysis.54 The activated procoagulant mechanism produces thrombin activatable fibrinolysis inhibitor (TAFI), which suppresses fibrinolysis.63. Tissue factor (TF), released into the plasma as a consequence of endothelial damage, activates PAI-1, which inhibits the formation of plasmin.62 Microthrombi cannot be removed; microvasculature and tissue perfusion is diminished and may cause ischaemia and hypoxia.62. The disruption to fibrinolysis is demonstrated by increased levels of the biomarker PAI-1.51 There is also evidence to suggest that the more marked the coagulation activation and fibrinolysis suppression, the greater the risk of mortality.51,64. TNF-, IL-1. disfunzione endoteliale. TAFI: tissue activatable fibrinolysis inhibitor, PAI-1: plasminogen activation inhibitor-1, TNF: tumour necrosis factor, IL: interleukin.")
20
Other factors involved in the host response
Risposta dell’ospite Patogeno Infezione Other factors Inflammation Endothelial dysfunction Coagulation/ fibrinolysis perdita omeostasi disfunz. organo morte

21
Ossido nitrico e sepsi SEPSI Ossido nitrico Inibizione respirazione
mitocondriale Cell signalling Ridotta espressione di P-selectin Ossido nitrico Altri effetti fisiologici Inibisce la proliferazione delle cellule muscolari lisce Stimola angiogenesi Effetti citotossici Nitric oxide (NO) has a number of important regulatory roles under resting conditions.67,68,69 In sepsis and the ensuing inflammation, NO mediates a number of pathophysiological changes, which include its cytotoxic function in infection. This increased synthesis of NO in response to infection may also lead to excessive vasodilatation, vascular hyporeactivity to vasoconstrictor agents and depression of myocardial contractility which may, in turn, lead to septic shock.70 iporeattività vascolare e vasodilatazione Ridotta adesione leucocitaria Inibizione di adesione/ aggregaz. piastrinica Shock settico
 has a number of important regulatory roles under resting conditions.67,68,69. In sepsis and the ensuing inflammation, NO mediates a number of pathophysiological changes, which include its cytotoxic function in infection. This increased synthesis of NO in response to infection may also lead to excessive vasodilatation, vascular hyporeactivity to vasoconstrictor agents and depression of myocardial contractility which may, in turn, lead to septic shock.70. iporeattività vascolare e vasodilatazione. Ridotta adesione. leucocitaria. Inibizione di adesione/ aggregaz. piastrinica. Shock settico.")
22
Il ruolo dell’ossido nitrico (NO) effetti vasomotori
Stimolo (e.g. acetylcholine/shear stress) Vasodilatazione/ regolazione del flusso ematico cellula tessutale ecNOS Ossido Nitrico Eccessiva vasodilatazione Ipotensione Refrattaria iNOS Stimolo (e.g. LPS, IL-1, TNF, PAF) Sepsi Over-expression of iNOS In sepsis and severe sepsis, overproduction of nitric oxide (NO) plays a key role in the inflammation cascade and can lead to septic shock. Inducible nitric oxide synthase (iNOS) expression is a slower process compared with endothelial constitutive nitric oxide synthase (ecNOS) expression and is observed mainly in conditions of infection and inflammation, such as sepsis and severe sepsis.71 In sepsis, NO production becomes abnormally elevated and it plays a key role in the inflammatory cascade and subsequent severe sepsis and septic shock.67,68,71 ecNOS function is decreased and endotoxins and pro-inflammatory cytokines stimulate expression of iNOS, which, unlike ecNOS, produces large amounts of NO for prolonged periods.67 This overproduction of NO causes mitochondrial inhibition43 and also severe vasodilatation and decreased vasopressor responsiveness, which can lead to refractory hypotension – the main clinical feature of septic shock.10,68 SHOCK SETTICO ecNOS: endothelial constitutive nitric oxide synthase, iNOS: inducible nitric oxide synthase LPS: lipopolysaccharide, IL: interleukin, PAF: platelet activating factor, TNF: tumour necrosis factor
 Vasodilatazione/ regolazione del flusso ematico. cellula. tessutale. ecNOS. Ossido Nitrico. Eccessiva vasodilatazione. Ipotensione Refrattaria. iNOS. Stimolo. (e.g. LPS, IL-1, TNF, PAF) Sepsi. Over-expression. of iNOS. In sepsis and severe sepsis, overproduction of nitric oxide (NO) plays a key role in the inflammation cascade and can lead to septic shock. Inducible nitric oxide synthase (iNOS) expression is a slower process compared with endothelial constitutive nitric oxide synthase (ecNOS) expression and is observed mainly in conditions of infection and inflammation, such as sepsis and severe sepsis.71. In sepsis, NO production becomes abnormally elevated and it plays a key role in the inflammatory cascade and subsequent severe sepsis and septic shock.67,68,71 ecNOS function is decreased and endotoxins and pro-inflammatory cytokines stimulate expression of iNOS, which, unlike ecNOS, produces large amounts of NO for prolonged periods.67 This overproduction of NO causes mitochondrial inhibition43 and also severe vasodilatation and decreased vasopressor responsiveness, which can lead to refractory hypotension – the main clinical feature of septic shock.10,68. SHOCK SETTICO. ecNOS: endothelial constitutive nitric oxide synthase, iNOS: inducible nitric oxide synthase. LPS: lipopolysaccharide, IL: interleukin, PAF: platelet activating factor, TNF: tumour necrosis factor.")
23
Lattati Sono un marker di ipossia tessutale precoce
La loro comparsa precede il danno d’organo > 36 mg/dl




















>")

