Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Telethon Institute of Genetics and Medicine, Napoli
Cromosomopatie Vincenzo Nigro Laboratorio di genetica - Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine, Napoli

2
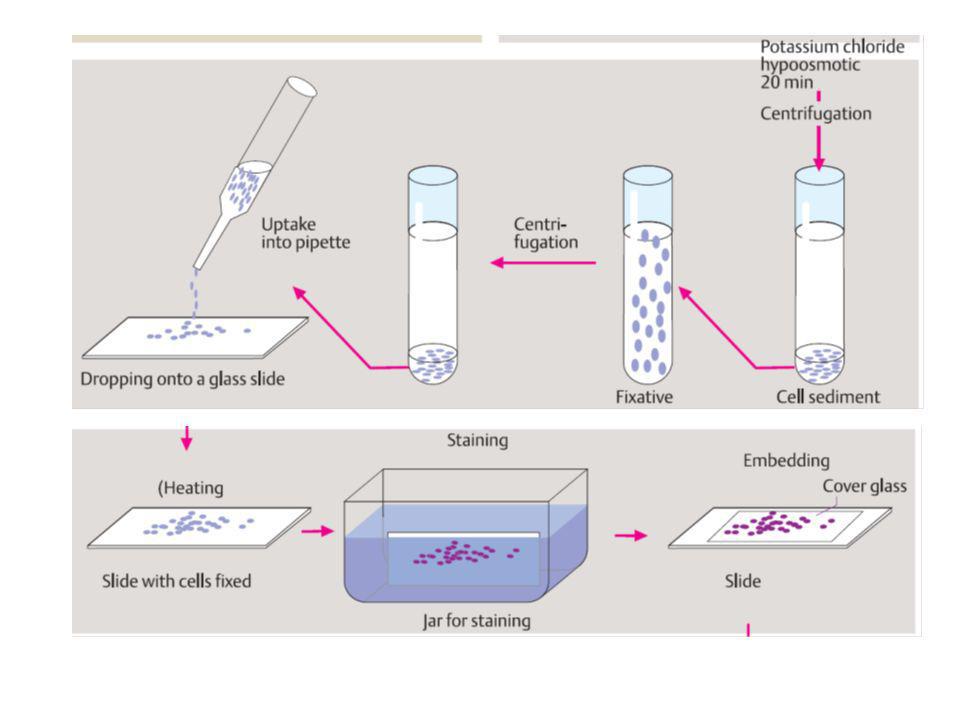
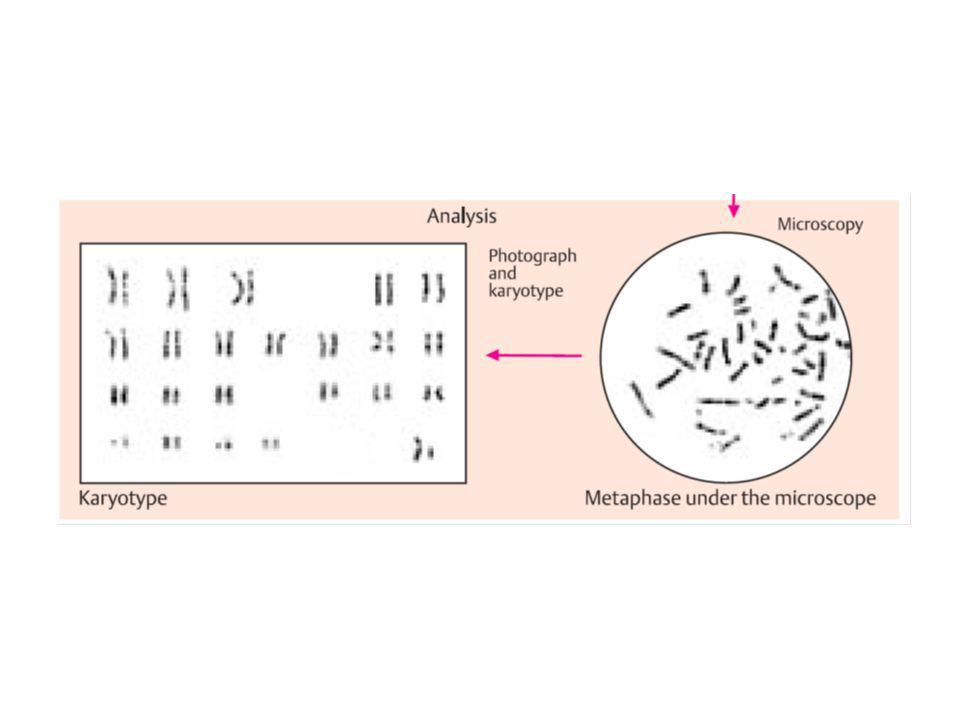
citogenetica di routine
da linfociti sono rappresentativi di ciascun altra cellula del corpo

5
metacentrici, se il centromero è centrale 1, 2, 3, 16, 17, 18, 19
submetacentrici, se il centromero non è centrale e non è vicino ad un’estremità 4, 5, 6, 7, 8, 9, 10, 11, 12, 20, X, Y acrocentrici, se il centromero è vicino ad un’estremità 13, 14, 15, 21, 22

6
Eteromorfismi citogenetici
Variazione pericentromerica del crom. 9 9qh+ Inversione 9 inv Variazione + inversione

7
Ereditarietà della variazione pericentromerica del cromosoma 1

8
citogenetica prenatale
da amniociti da villi coriali dovrebbero essere rappresentativi delle cellule del feto difficili da ottenere

9
Frequenza di anomalie cromosomiche negli aborti spontanei (39. 8%-40
trisomie autosomiche 49-52% Turner (45, X) 15-19% triploidia (69) 15-16% tetraploidia (92) 5-6% altre anomalie 6-14%
 15-19% triploidia (69) 15-16% tetraploidia (92) 5-6% altre anomalie 6-14%")
10
Le alterazioni cromosomiche sono più frequenti al crescere dell’età materna, mentre le mutazioni puntiformi sono legate al numero di divisioni cellulari che avvengono circa ogni 15 gg nella linea germinale maschile

11
donna di 37 anni e mezzo alla data presunta del parto
MESI COMPLETATI ANNI Rischio di 1/239 di figlio con sindrome di Down

12
aneuploidie nei nati vivi
anomalie autosomiche trisomia 21 (sindrome di Down) 1/700 trisomia 18 (sindrome di Edwards) 1/6.500 trisomia 13 (sindrome di Patau) 1/20.000 anomalie dei cromosomi sessuali Sindrome di Klinefelter (47,XXY) 1/900 maschi Sindrome XYY (47,XYY) 1/1.000 maschi Sindrome della tripla X (47,XXX) 1/1.200 femmine Monosomia X o S. di Turner (45,X0) 1/2.500 femmine
 1/700. trisomia 18 (sindrome di Edwards) 1/ trisomia 13 (sindrome di Patau) 1/ anomalie dei cromosomi sessuali. Sindrome di Klinefelter (47,XXY) 1/900 maschi. Sindrome XYY (47,XYY) 1/1.000 maschi. Sindrome della tripla X (47,XXX) 1/1.200 femmine. Monosomia X o S. di Turner (45,X0) 1/2.500 femmine.")
13
anomalie ecografiche maggiori

14
anomalie ecografiche minori
“soft markers”

15
tritest interpretazione dei risultati
anomalia fetale AFP Alfa-feto proteina Beta hCG uE estriolo non coniugato NTD =difetti del tubo neurale* Normal Trisomia 21 18 * NTD: anencefalia, spina bifida and encefalocele

16
Il duotest (double screen) include la valutazione del PAPP-A (Pregnancy Associated Plasma Protein A) e la frazione libera della gonadotropina corionica (free-betaHCG). Viene effettuato tra la 10ma e la 13ma settimana di gravidanza dal siero della gestante Translucenza nucale free-bHCG PAPP-A Trisomia 21 ++ - Trisomia 13,18 +++ - - S. di Turner ++++ +/- Triploidia materna - - - Triploidia paterna

17
trisomia 21 Down Il 70% delle gravidanze non giunge a termine

18
1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 Meiosis 1 Meiosis 1 error Meiosis 1 Meiosis 1 1 1 1 1 2 2 1 1 1 1 + 1 2 1 2 1 2 1 2 1 2 Meiosis 2 Meiosis 2 error Meiosis 2 Meiosis 2 1 1 1 2 1 1 1 or + or 3 other combinations + or other combinations Mitotic error 1 2 1 1 2 2 1 1 1 2 2 SEA3069

19
origine dell’extra cromosoma 21
MM2 19.8% PM1 2.6% MM1 68% PM2 4.1% MIT 5.5% Data from the Antonarakis and Hassold laboratories sea3109

20
anomalie cromosomiche nella sindrome di Down
13 12 11.2 p 11.1 27 11.1 mosaicism 11.2 2 altre t 3 t21;22 17 21 t21;21 5 Anomalia t15;21 q 6 t13;21 15 22.1 t14;21 925 free T21 D21S17 22.2 DSCR 1 ETS2 10 100 1000 22.3 Numero MX1 HC21

21
Ipotesi sulla variabilità del fenotipo di 6 individui diversi in caso di trisomia 21 in presenza di varianti alleliche fenotipo Livello di espressione

22
trisomia 21 Down 40.000 casi in Italia Neurologici :
Ritardo mentale 100% Alzheimer dopo i 35anni 100% Ipotonia muscolare 100% Bassa statura 70% Testa : Brachicefalia 75% Epicanto 60% brushfield spots iride 55% lingua protrudente 45% orecchie displastiche 50%

23
trisomia 21 Down Arti corti, mani larghe 65% Mignolo corto 60%
Solco palmare trasverso 60% Cuore Difetti cardiaci congeniti 40% Canale atrioventricolare 16% Anomali gastrointestinali Atresia/stenosi duodenale 250x ano imperforato 50x Hirschsprung 300x Sangue: Leucemia acuta megacariocitica 300x Leucemia (ALL e AML) x
 10-20x.")
24
trisomia 18 Edwards (1/6.500 nati)
90% dei casi nondisgiunzione materna M/F = 1/4 Giunge a termine solo il 2.5% dei concepimenti Di questi il 33% muore nel primo mese, il 50% entro 2 mesi Oltre 100 anomalie Peso sotto la norma, difficoltà suzione Ipotonia Idrocefalo, epilessia Malformazioni cardiache sinclinodattilia, unghie poco sviluppate piedi a calcagno prominente Gambe incrociate

25
trisomia 13 Patau (1/12.000-20.000 nati)
(1/ nati) 90% dei casi nondisgiunzione materna Giunge a termine solo il 2.5% dei concepimenti Di questi il 33% muore nel primo mese, il 50% entro 2 mesi Peso sotto la norma, difficoltà suzione Oloprosencefalia, microcefalia Cecità e sordità Occhi che possono fondersi Labiopalatoschisi 80% epilessia Malformazioni cardiache sinclinodattilia piedi a calcagno prominente
 90% dei casi nondisgiunzione materna. Giunge a termine solo il 2.5% dei concepimenti. Di questi il 33% muore nel primo mese, il 50% entro 2 mesi. Peso sotto la norma, difficoltà suzione. Oloprosencefalia, microcefalia. Cecità e sordità. Occhi che possono fondersi. Labiopalatoschisi 80% epilessia. Malformazioni cardiache. sinclinodattilia. piedi a calcagno prominente.")
26
Monosomia X (45,X0) Turner 1:2.500
Solo l’1% delle gravidanze giunge a termine Errore nella spermatogenesi nell’ 80% dei casi e non correla con l’età dei genitori Caratteristiche principali: mancato sviluppo ovarico con amenorrea primaria e sterilità fenotipo molto variabile linfedema con rigonfiamento delle mani e dei piedi pterigio del collo

27
Monosomia X (45,X0) Turner 1:2.500
mandibola più piccola (micrognazia) torace largo con aumento degli spazi intercostali attaccatura bassa delle orecchie bassa statura quarto metacarpo corto cardiopatia, ipertensione e anomalie renali. sia l’intelligenza sia l’attesa di vita sono normali
 torace largo con aumento degli spazi intercostali. attaccatura bassa delle orecchie. bassa statura. quarto metacarpo corto. cardiopatia, ipertensione e anomalie renali. sia l’intelligenza sia l’attesa di vita sono normali.")
28
feto con anomalia cromosomica (mosaicismo)
trisomie a mosaico 8, 9, 13, 18, 21 crescita in coltura di cellule materne mosaicismo vero (livello III) pseudomosaicismo (livelli II e I)
 pseudomosaicismo (livelli II e I)")
29
XX o XY Il sesso maschile è determinato dalla presenza del cromosoma Y
Si sono evoluti meccanismi per compensare la differenza di dosaggio genico del cromosoma X, presente in 2 copie nelle femmine e in 1 copia nei maschi

30
2 cromosomi X nelle donne,
1 solo negli uomini? il cromosoma X raddoppia l’espressione di tutti i geni contenuti, cioè si produce 2 volte più RNA nelle femmine uno dei due cromosomi X è inattivato casualmente in ciascuna cellula allo stadio di blastocisti

31
Il Klinefelter? Nel Klinefelter (XXY) uno dei due cromosomi X è inattivato casualmente in ciascuna cellula allo stadio di blastocisti Quindi il dosaggio sarebbe mantenuto

32
Sindrome di Klinefelter (47,XXY) 1:900-1:600 maschi
Il 50% delle gravidanze giunge a termine Fenotipo maschile Caratteristiche principali: Statura alta Ipogonadismo, bassi livelli di testosterone, mancata produzione di spermatozoi (azoospermia) e quindi sterilità Ginecomastia Sia l’intelligenza sia l’attesa di vita sono quasi normali
 e quindi sterilità. Ginecomastia. Sia l’intelligenza sia l’attesa di vita sono quasi normali.")
33
Altre forme citogenetiche
Ma ci sono anche Klinefeler 48,XXYY and 48,XXXY in 1 caso su 17,000 e 1 su 50,000 mnati maschi 49,XXXXY in 1 caso su 85, ,000 Ci sono maschi 46,XX in cui avviene una traslocazione di parte di cromosoma Y sul cromosoma X che include la sex determining region (SRY) mosaici
 mosaici.")
34
PAR1 ha 24 geni, PAR2 ha solo 4 geni
Le regioni PAR presenti sui cromosomi sessuali contengono geni che non sono inattivati, perché il doppio dosaggio è assicurato comunque PAR1 ha 24 geni, PAR2 ha solo 4 geni

35
Il gene SHOX Short stature HOmeoboX-containing
Mutazioni o delezioni del gene SHOX nella regione PAR1 causa ritardo di crescita e bassa statura. La bassa statura di donne con sindrome di Turner Syndrome (X0) è il risultato di una sola copia di SHOX (ma anche il quarto metacarpo corto) La maggiore statura nel Klinefelter (XXY) e nella tripla X (XXX) potrebbe essere il risultato di 3 copie di SHOX
 è il risultato di una sola copia di SHOX (ma anche il quarto metacarpo corto) La maggiore statura nel Klinefelter (XXY) e nella tripla X (XXX) potrebbe essere il risultato di 3 copie di SHOX.")
36
3 copie nel Klinefelter, ma anche nella tripla X
variabilità dei geni del cromosoma X delle regioni PAR, quindi non inattivati 3 copie nel Klinefelter, ma anche nella tripla X Mario Rossi Luca Bianchi Pio Verdi Giulio Rosa Lucio Viola Gianni Neri Livello di espressione

37
A complicare le cose… circa il 15 % dei geni umani presenti sull’X sfugge all’inattivazione, mentre nel topo questa è un’evenienza rara (solo 6 geni in tutto) alcuni sono espressi al % altri al 10% questo fenomeno è quindi incompleto e le donne hanno una elevata eterogeneità nell’espressione di geni dell’X
 alcuni sono espressi al % altri al 10% questo fenomeno è quindi incompleto e le donne hanno una elevata eterogeneità nell’espressione di geni dell’X.")
38
Manifestazione clinica: NO Manifestazione clinica: SI
ipotesi sulla variabilità di ogni singola manifestazione clinica di Klinefelter in presenza di varianti in geni del cromosoma X non inattivati Manifestazione clinica: NO Manifestazione clinica: SI Mario Rossi Luca Bianchi Pio Verdi Giulio Rosa Lucio Viola Gianni Neri Livello di espressione

39
Quanti Klinefelter? Prevalenza di XXYs è cresciuta da 1.09 a 1.72 per 1000 maschi nati (P=0.023) Questo incremento non è dovuto all’aumento dell’età materna Sono nati maschi in Italia e maschi in Campania nel 2007 max nuovi Klinefelter ogni anno in Italia (32-52 in Campania)
")
40
XXY è la sola trisomia nota in cui circa il 50% dei casi è causato da una non disgiunzione alla prima divisione meiotica paterna

41
Trisomia X (47,XXX) 1:1.200 Il 70% delle gravidanze giunge a termine
Errore nella disgiunzione materna e correla con l’età materna Caratteristiche principali: Statura alta Fertilità normale, irregolarità ciclo Sia l’intelligenza sia l’attesa di vita sono normali

42
Maschio (47,XYY) 1:1.000 maschi Fenotipo maschile
Caratteristiche principali: Statura alta Fertilità normale Non vi è correlazione con l’età paterna Sia l’intelligenza sia l’attesa di vita sono perfettamente normali

43
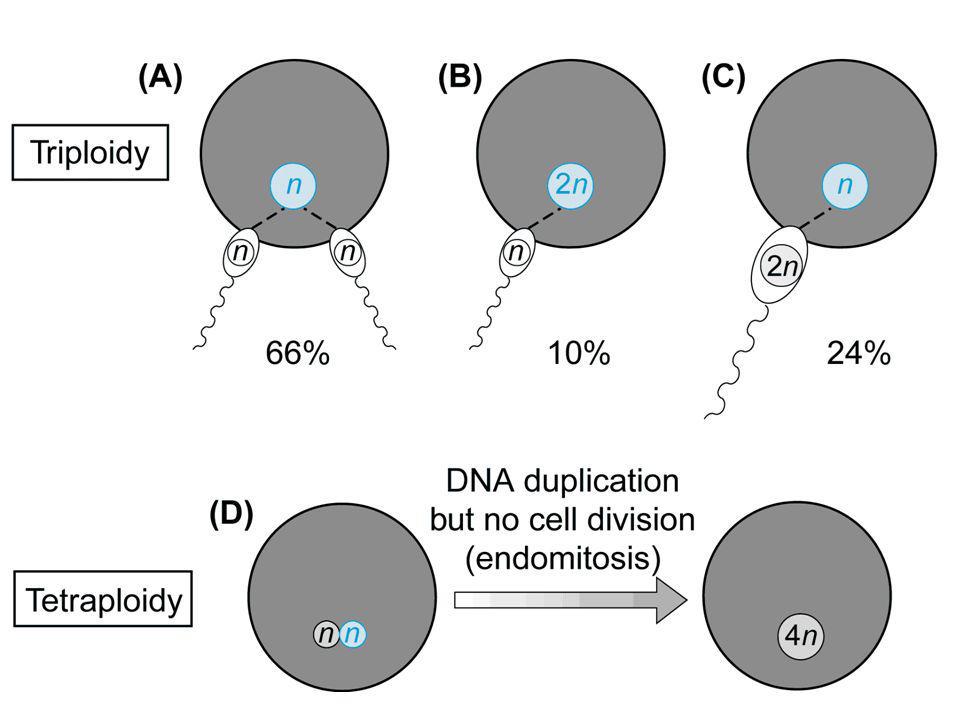
triploidia Frequenza alla nascita = 1/10.000
Frequenza negli aborti = 1/14 Cariotipo 69,XXY 57% Cariotipo 69,XXX 40% Cariotipo 69,XYY 3%

45
Nati vivi Tipo I, corredo sovrannumerario paterno
Feto microcefalico o normale Placenta ingrossata Tipo II, corredo sovrannumerario materno Ritardo di crescita Feto con macrocefalia relativa Placenta poco sviluppata Nati vivi Basso peso Asimmetria cranio-facciale e difetti di ossificazione del cranio Microftalmia, ipertelorismo, micrognazia Sindattilia cutanea, piedi torti Anomalie genitali, ipoplasia delle surrenali Cardiopatie

46
Mosaicismo, quando il cambiamento avviene dopo che si è formato lo zigote
47,XXY/46,XY

47
Rischio riproduttivo generale
per una coppia per cui l’anamnesi personale e familiare abbiano escluso un incremento del rischio rispetto alla popolazione è 3-5% in caso di difetti congeniti rilevabili alla nascita (a. crom 0.65%) 8-10% rilevabili entro i 10 anni di età
 8-10% rilevabili entro i 10 anni di età.")
48
Un precedente figlio con anomalie cromosomiche
Aumenta il rischio in caso di: tutte le trisomie non mosaico riarrangiamenti strutturali marker cromosomi

49
Un precedente figlio con anomalie cromosomiche
NON aumenta il rischio in caso di: 47, XYY triploidia, tetraploidia sindrome di Turner

Presentazioni simili