Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
mutazioni puntiformi Vincenzo Nigro Dipartimento di Patologia Generale Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine (TIGEM)
")
2
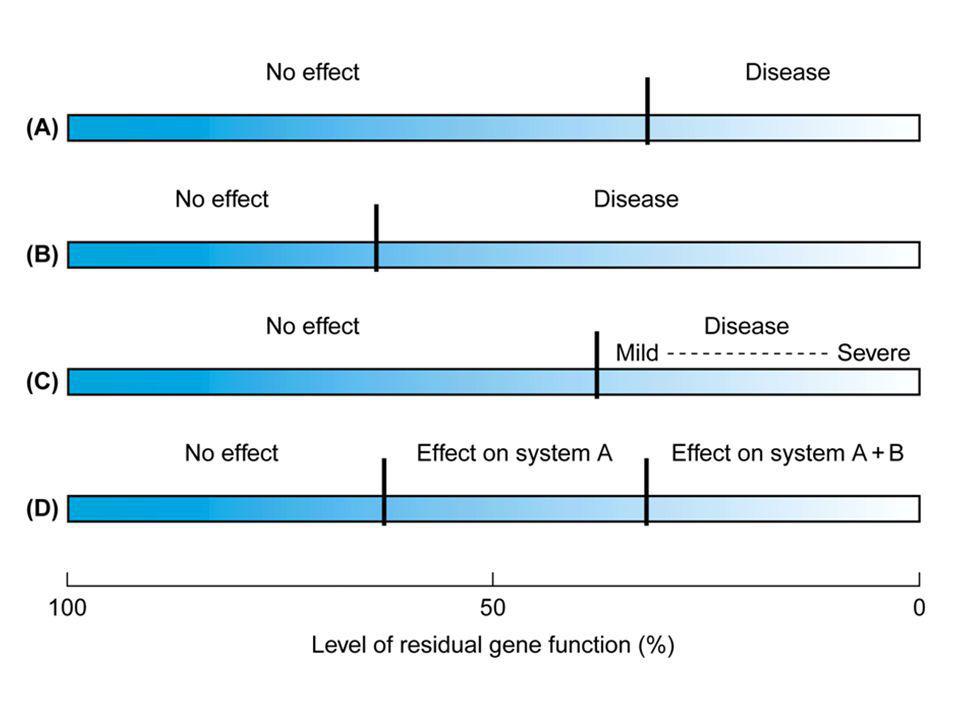
5 effetti di un allele nullo o amorfo = nessun prodotto genico ipomorfo = ridotta quantità/attività ipermorfo = aumentata quantità/attività neomorfo = nuova quantità/attività antimorfo = quantità/attività antagonistica (dominante negativo)
")
3
mutazioni puntiformi missenso Le mutazioni missenso sono quelle in cui il cambiamento determina nel prodotto proteico la sostituzione di un aminoacido con un aminoacido differente Sebbene queste alterazioni generalmente non provochino conseguenze nella funzionalità della proteina (polimorfismi o varianti), ci sono casi in cui anche una minima alterazione può avere conseguenze gravi.
, ci sono casi in cui anche una minima alterazione può avere conseguenze gravi.")
5
Mutazioni dei codoni umani (mutazioni independenti nei geni F8, F9, L1CAM, OTC, BTK)
")
6
mutazioni puntiformi nonsenso La mutazione nonsenso è quella in cui la modificazione nucleotidica provoca la creazione di un tripletta di stop, che blocca la sintesi della proteina prematuramente. In questo caso, la funzionalità della proteina dipenderà dalla posizione dello stop.

7
mutazioni nonsenso SEA 3063 UGA UAAUAG Su 731 mutazioni independenti in 9 patologie del cromosoma X

8
mutazioni frame-shift Le mutazioni frame-shift o di slittamento del modulo di lettura consistono nellinserzione o delezione di un numero di nucleotidi non divisibile per 3 (1, 2, 4, 5, 7, 8, 10, ecc.) con conseguente sfasamento della cornice di lettura delle triplette dell'RNA messaggero. Questa mutazione determina la traduzione non corretta della proteina a valle della mutazione.

9
Numerazione dei nucleotidi Nucleotidi del cDNA Il nucleotide +1 è la A dell ATG-codone di inizio della traduzione Il nucleotide che precede al 5' lATG-codone di inizio della traduzione è denominato -1; non esiste una base 0 Il nucleotide che segue al 3' il codone di terminazione è denominato *1

10
Nucleotidi intronici fiancheggianti inizio dellintrone: il numero dellultimo nucleotide dellesone che precede, il segno + e la posizione nellintrone, ad esempio 77+1G, 77+2T, oppure quando il numero dellesone è noto ed univoco IVS1+1G, IVS1+2T fine dellintrone: il numero del primo nucleotide del esone seguente, il segno – e la posizione a monte dellesone, ad esempio 78-2A, 78-1G, oppure quando il numero dellesone è noto ed univoco, IVS1-2A, IVS1-2G

11
sostituzioni le sostituzioni sono indicate dal carattere >. Ad esempio, 76A>C indica che in posizione 76 unadenina è sostituita da una citosina 88+1G>T (oppure IVS2+1G>T) indica che una guanina è sostituita da una timina in posizione +1 dellintrone 2, posizionato tra i nucleotidi 88 e 89 del cDNA 89-2A>C (oppure IVS2-2A>C) indica che unadenina è sostituita da una citosina in posizione -2 dellintrone 2, posizionato tra i nucleotidi 88 e 89 del cDNA
 indica che una guanina è sostituita da una timina in posizione +1 dellintrone 2, posizionato tra i nucleotidi 88 e 89 del cDNA 89-2A>C (oppure IVS2-2A>C) indica che unadenina è sostituita da una citosina in posizione -2 dellintrone 2, posizionato tra i nucleotidi 88 e 89 del cDNA.")
13
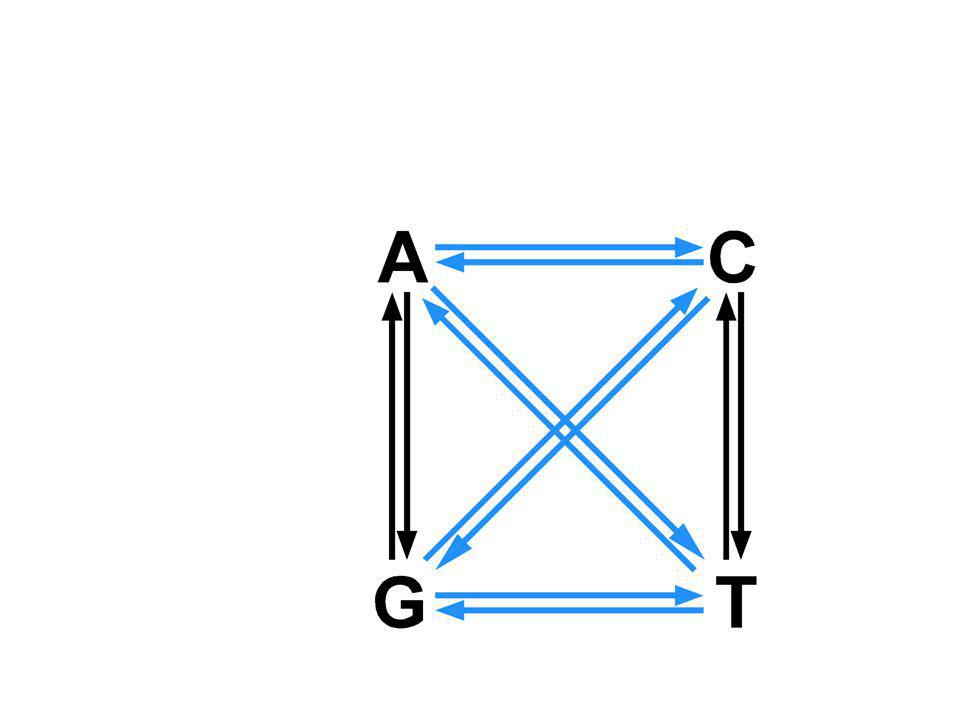
mutazioni teoricamente previste SEA 3057 trasversioni T>C, C>T, G>A, A>G T>A o G, C>G or A G>T o C, A>T or G transizioni SNPs 12,000,000 46,000 transizioni trasversioni

14
NH 2 N N N N N NH OO H3CH3CH3CH3C Cytosine 5-methylcytosineThymine metilazionedeaminazione O O CG TG CG CA Il meccanismo più comune di mutazione

16
eredità autosomica dominante a penetranza completa

17
acrocefalosindattilia sindrome di Apert 1:65.000 alla nascita craniosinostosi, volta cranica a forma conica ipertensione endocranica ritardo mentale ipoplasia della parte centrale della faccia sindattilia delle dita delle mani e dei piedi sordità e atrofia ottica

18
acrocefalosindattilia sindrome di Apert tutti i pazienti hanno la stessa mutazione Apert (Cys755Gly) del gene human fibroblast growth factor receptor 2 (FGFR2) la mutazione è in eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Crouzon e Pfeiffer
 del gene human fibroblast growth factor receptor 2 (FGFR2) la mutazione è in eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Crouzon e Pfeiffer")
19
sindrome di Pfeiffer alcuni pazienti hanno la mutazione Pfeiffer (Cys342Arg) del gene human fibroblast growth factor receptor 2 (FGFR2) altri la mutazione Pro252Arg in FGFR1 la mutazione è in eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Crouzon e Apert
 del gene human fibroblast growth factor receptor 2 (FGFR2) altri la mutazione Pro252Arg in FGFR1 la mutazione è in eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Crouzon e Apert")
20
disostosi cranio facciale sindrome di Crouzon alcuni pazienti hanno la mutazione (Cys342Tyr) del gene human fibroblast growth factor receptor 2 (FGFR2) la mutazione è in eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Pfeiffer e Apert con alcune mutazioni in comune
 del gene human fibroblast growth factor receptor 2 (FGFR2) la mutazione è in eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Pfeiffer e Apert con alcune mutazioni in comune")
21
acondroplasia nanismo dismorfico (1:35.000) arti corti e testa sproporzionatamente più grossa fronte prominente e naso appiattito altezza media 130 cm nei maschi 125 cm nelle femmine La mutazione è in eterozigosi Gly380Arg nel recettore 3 del "fibroblast growth factor" (FGFR3) a 4p16.3 autosomico dominante a penetranza completa
 arti corti e testa sproporzionatamente più grossa fronte prominente e naso appiattito altezza media 130 cm nei maschi 125 cm nelle femmine La mutazione è in eterozigosi Gly380Arg nel recettore 3 del fibroblast growth factor (FGFR3) a 4p16.3 autosomico dominante a penetranza completa")
22
acondroplasia La mutazione conferisce una funzione aumentata al recettore dell'FGF (allele ipermorfo) che è una tirosin- chinasi di membrana In risposta all'FGF il recettore dimerizza e si fosforila trasducendo un segnale con la funzione di rallentare la proliferazione dei condrociti e quindi la crescita ossea Topi senza il gene FGF3R hanno ossa lunghe e vertebre allungate
 che è una tirosin- chinasi di membrana In risposta all FGF il recettore dimerizza e si fosforila trasducendo un segnale con la funzione di rallentare la proliferazione dei condrociti e quindi la crescita ossea Topi senza il gene FGF3R hanno ossa lunghe e vertebre allungate")
24
ipocondroplasia L'ipocondroplasia ha caratteristiche simili all'acondroplasia, ma di gravità minore con un coinvolgimento craniofacciale inferiore. L'altezza può risultare ai limiti della norma e la malattia viene spesso non diagnosticata. L'ipocondroplasia è meno omogenea: circa il 70% dei casi è dovuto alla sostituzione N540K del gene FGFR3, mentre non si conosce la mutazione nel restante 30%.

25
frequenza relativa di mutazioni de novo che causano acondroplasia i rapporto alletà paterna

26
numero di divisioni nelle linea germinale maschile

28
mutazioni eterozigoti di PAX3 Waardenburg

29
sordità (o deficit uditivo di vario livello) bilaterale,sordità modifiche nella pigmentazione, sia dei capelli (albinismo parziale, in genere piebaldismo) che della pelle,capellipelle anomalie nello sviluppo dei tessuti derivati dalla cresta neurale lateralizzazione del canto mediale diverso colore degli occhi (eterocromia), di solito uno marrone e l'altro blueterocromia
 bilaterale,sordità modifiche nella pigmentazione, sia dei capelli (albinismo parziale, in genere piebaldismo) che della pelle,capellipelle anomalie nello sviluppo dei tessuti derivati dalla cresta neurale lateralizzazione del canto mediale diverso colore degli occhi (eterocromia), di solito uno marrone e l altro blueterocromia")
30
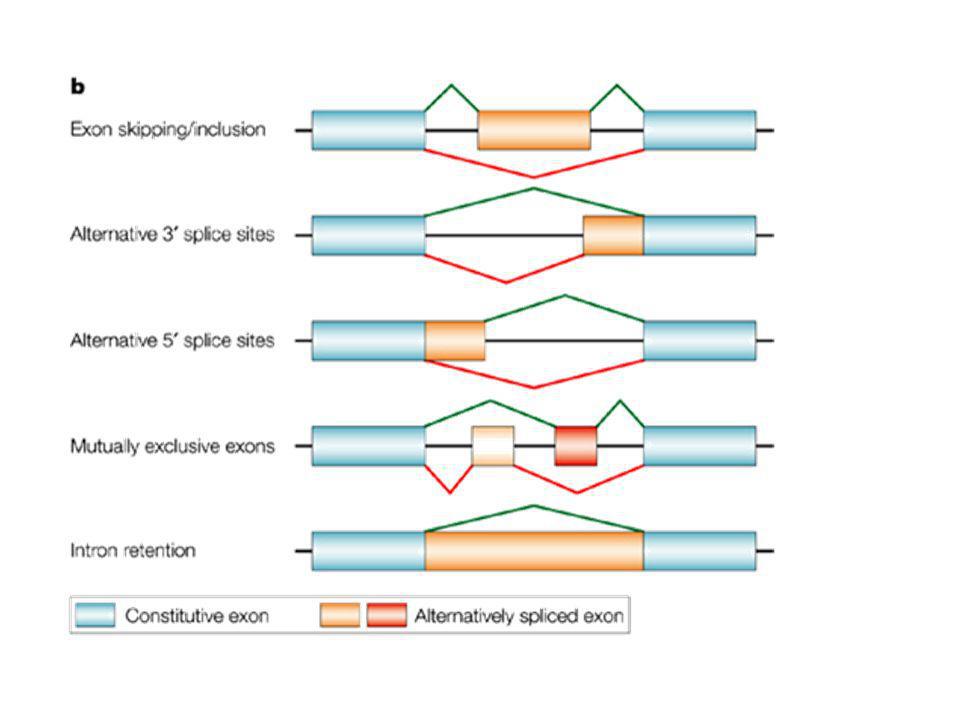
Motivi classici di splicing

32
HGMD Mutations in Intron splice sites (5Jan07) 3498 Donor Splice Site mutations 2296 Acceptor Splice Site mutations
 3498 Donor Splice Site mutations 2296 Acceptor Splice Site mutations")
33
Splicing Abnormalities exonexonexonIVSIVS 5ss5ss3ss3ss Normal 5ss mutation; exon skipping 3ss mutation; exon skipping 5ss mutation; use of cryptic 5ss 3ss mutation; use of cryptic 3ss Activation of cryptic 5ss Activation of cryptic 5ss and use of cryptic 3ss Splicing enhancer mutation Lariat structure branchpoint mutation

34
Progeria Hutchinson-Gilford invecchiamento precoce bassa statura, pelle rugosa calvizie, assenza di tessuto adiposo aterosclerosi ed infarto

35
Progeria Hutchinson-Gilford nuova mutazione in eterozigosi del gene lamina A la mutazione è in eterozigosi de novo cromosoma 1q23 La mutazione non cambia l'aminoacido glicina G608G, ma introduce un sito donor di splicing GGT che fa perdere 50 aminoacidi alla proteina sperimentazione con inibitori di farnesil-trasferasi

37
conversione genica

38
DHPLC denaturing high-performance liquid chromatography Serve a stabilire se, se un frammento di DNA è identico ad un frammento standard o contiene uno o più nucleotidi diversi E una tecnica in HPLC a fase inversa in cui una fase stazionaria costituita da microsferette omogenee di polistirene divinilbenzene è in grado di trattenere una molecola polare come il DNA grazie allazione di un controione, rappresentato dal Trietil Ammonio Acetato (TEAA 0.1M) La parte polare del TEAA interagisce con il DNA e la parte apolare con la fase stazionaria Essa si basa sui differenti tempi di eluizione applicando un gradiente di acetonitrile di eteroduplex e omoduplex DNA Il DNA omoduplex è quello nativo, mentre leteroduplex si forma quando sono denaturati e rinaturati nella stessa provetta due frammenti di DNA che hanno una piccola differenza nella sequenza di basi
 La parte polare del TEAA interagisce con il DNA e la parte apolare con la fase stazionaria Essa si basa sui differenti tempi di eluizione applicando un gradiente di acetonitrile di eteroduplex e omoduplex DNA Il DNA omoduplex è quello nativo, mentre leteroduplex si forma quando sono denaturati e rinaturati nella stessa provetta due frammenti di DNA che hanno una piccola differenza nella sequenza di basi")
39
DHPLC CGTA AlleleA B TACG GCAT Denaturazionee raffreddamento MUT WT Heteroduplex Homoduplex A B C

Presentazioni simili















 DI BREVI TRATTI RIPETUTI>")






