Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

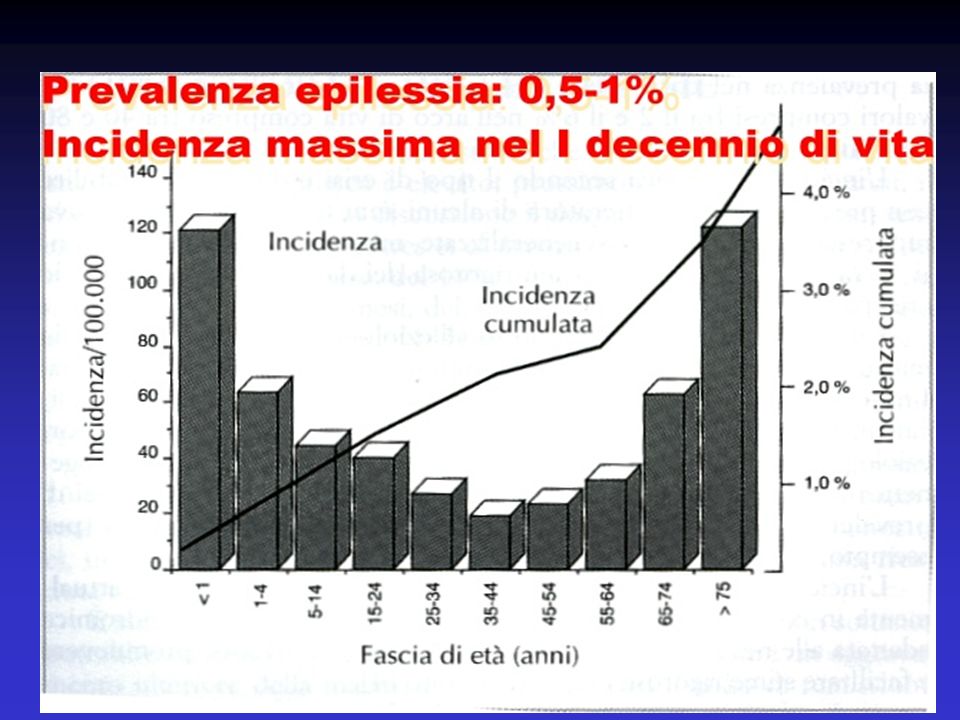
1
EPILESSIA

2
COS’E’ UNA CRISI EPILETTICA?
Evento clinico accessuale , a semeiologia proteiforme ma stereotipa nella sua presentazione intrasoggetto, dovuto ad una scarica neuronale abnorme COS’E’L’EPILESSIA Sindrome clinica, eterogenea per eziologia e presentazione, caratterizzata dalla RIPETIZIONE di crisi epilettiche NON SINTOMATICHE di una lesione ACUTA del sistema nervoso

3
Epilessia: definizioni.
Crisi sintomatica acuta. Conseguente ad evidente evento patologico immediatamente precedente; può essere isolata. Crisi sintomatica remota. Conseguente a pregressa lesione cerebrale. Stato di male epilettico. Le crisi sono continue o si ripetono senza il ritorno di una funzionalità neurologica normale tra le singole crisi.

5
Classificazione delle crisi (ILAE, 1981)
Assenze tipiche Assenze atipiche Crisi miocloniche Crisi toniche Crisi atoniche Crisi tonico-cloniche crisi generalizzate Semplici Complesse Con generalizzazione secondaria crisi parziali

6
CRISI GENERALIZZATE La scarica parossistica interessa fino dall’esordio i due emisferi cerebrali e sembra coinvolgere contemporaneamente l’insieme della corteccia cerebrale Clinicamente, non vi sono segni o sintomi che facciano pensare ad una partenza in un sistema anatomico o funzionale localizzato Le manifestazioni motorie, quando presenti, sono bilaterali

7
ASSENZE: CRISI DI BREVE DURATA, LA CUI MANIFESTAZIONE PRINCIPALE E’ UNA ALTERAZIONE DELLA COSCIENZA
TIPICHE Esordio e fine bruschi Complessi punta-onda a 3 Hz diffusi all’EEG Osservabili in epilessie generalizzate idiopatiche ATIPICHE Esordio e fine più progressivi e meno differenziabili Alterazione della coscienza in genere meno profonda Osservabili in epilessie sintomatiche dell’infanzia

8
MIOCLONIE Contrazioni muscolari simultanee di agonisti ed antagonisti
Brevi, con tipografia ed intensità variabile, talora massive (con caduta), talora parcellari Tipiche di alcune epilessie generalizzate, sia idiopatiche (sindrome di Janz), che sintomatiche (epilessia miocloniche progressive)
, talora parcellari. Tipiche di alcune epilessie generalizzate, sia idiopatiche (sindrome di Janz), che sintomatiche (epilessia miocloniche progressive)")
9
CRISI TONICHE Contrazione della muscolatura assiale o di tutta la muscolatura, della durata di alcuni secondi Associate solitamente ad alterazione della coscienza ed a manifestazioni vegetative Si osservano in epilessie sintomatiche (Lennox-Gastaut)
")
10
CRISI ATONICHE Improvvisa risoluzione del tono muscolare, con caduta violenta e traumatizzante Talora parcellari (caduta del capo) Osservabili in epilessie sintomatiche Talora associata a mioclono (crisi mioclono-astatica)
")
11
CRISI TONICO-CLONICA (GRANDE MALE)
Si manifestano senza prodromi, con tre fasi Fase tonica (10-20 secondi): perdita di coscienza, spesso preceduta da grido, contrazione sostenuta di tutta la muscolatura, può esserci morsus Fase clonica: Progressiva comparsa di scosse muscolari bilaterali e simmetriche, ritmiche, di ampiezza progressivamente maggiore Fase post-critica: coma, ipotonia, progressiva ripresa della coscienza
: perdita di coscienza, spesso preceduta da grido, contrazione sostenuta di tutta la muscolatura, può esserci morsus. Fase clonica: Progressiva comparsa di scosse muscolari bilaterali e simmetriche, ritmiche, di ampiezza progressivamente maggiore. Fase post-critica: coma, ipotonia, progressiva ripresa della coscienza.")
12
CRISI PARZIALI La scarica parossistica interessa inizialmente un settore limitato della corteccia cerebrale (ZONA EPILETTOGENA) La semeiologia clinica delle crisi parziali dipende strettamente e direttamente dalle caratteristiche anatomofunzionali del circuito epilettogeno, e cioè dall’area di partenza (importanza del sintomo iniziale) e dalle strutture via via reclutate
 e dalle strutture via via reclutate.")
13
CRISI PARZIALI MOTORIE Cloniche Versive
Posturali fonatorie SENSITIVE -Somatosensitive -Visive -Uditive -Gustative -olfattive VEGETATIVE PSICHICHE SEMPLICI: coscienza integra, il paziente ricorda la crisi e la può descrivere COMPLESSE:rottura del contatto con l’ambiente, spesso associata ad automatismi di vario tipo, coperte da amnesia parziale o totale

14
SIGNIFICATO LOCALIZZATORIO DELLE CRISI PARZIALI
MOTORIE Area rolandica controlaterale VERSIVE Area 8 oculogira, area supplementare POSTURALI Area 6 ANARTRICHE Piede 3 circonvoluzione frontale SOMATOSENSITIVE Aree postrolandiche controlaterali VISIVE Aree occipitali e temporali posteriori UDITIVE Prima circonvoluzione temporale OLFATTIVE Aree orbitofrontali, uncus temporale GUSTATIVE Lobo insula PSICHICHE Aree limbiche, necortex temporale AUTOMATISMI-OROALIMENTARI Aree limbiche, temporomesiali, amigdala AUTOMATISMI “BIZZARRI” Aree frontali

16
epilessia generalizzata
ILAE, 1989 Asse sintomatologico epilessia generalizzata idiopatica criptogenetica sintomatica Asse eziopatogenetico epilessia parziale idiopatica criptogenetica sintomatica

17
EPILESSIE GENERALIZZATE
Si presentano con crisi generalizzate EEG con segni epilettiformi generalizzati Presentazione età-dipendente Idiopatiche Geneticamente determinate Talora ereditarie Non legate ad apparenti lesioni strutturali Benigne (buona risposta alla terapia, non defcit aggiuntivi) Criptogenetiche/sintomatiche Legate a lesioni strutturali Associate ad altri segni neurologici
 Criptogenetiche/sintomatiche. Legate a lesioni strutturali. Associate ad altri segni neurologici.")
18
EPILESSIE GENERALIZZATE IDIOPATICHE (in ordine di età)
EPILESSIA MIOCLONICA BENIGNA EPILESSIA CON ASSENZE DELL’INFANZIA EPILESSIA CON ASSENZE DEL GIOVANE EPILESSIA MIOCLONICA GIOVANILE EPILESSIA CON GRANDE MALE AL RISVEGLIO

19
EPILESSIA CON ASSENZE DELL’INFANZIA
Esordio: 4-6 anni Numerosissime (anche >200) assenze tipiche al giorno, tipicamente scatenate dall’ iperventilazione EEG: normale attività di fondo, complessi punta-onda regolari a 3 Hz Non deficit neurologici associati Guarigione nel 50-60% dei casi con l’adolescenza Risposta a: VPA, ETS, LTG
 assenze tipiche al giorno, tipicamente scatenate dall’ iperventilazione. EEG: normale attività di fondo, complessi punta-onda regolari a 3 Hz. Non deficit neurologici associati. Guarigione nel 50-60% dei casi con l’adolescenza. Risposta a: VPA, ETS, LTG.")
20
EPILESSIA CON ASSENZE DEL GIOVANE
Esordio: 8-10 anni Assenze tipiche, meno frequenti rispetto alla forma infantile. Possibili scatti mioclonici e, più tardivamente, GTC EEG: normale attività di fondo, complessi punta-onda regolari a 3,5-4 Hz Non deficit neurologici associati Guarigione più rara, con frequenti recidive GTC in età più tardiva Risposta a: VPA, ETS, LTG

21
EPILESSIA MIOCLONICA GIOVANILE (sindrome di Janz)
Esordio: anni Mioclonie al mattino al risveglio, GTC. Possibili sporadiche assenze tipiche EEG: normale attività di fondo, complessi punta e polipunta-onda irregolari a 3,5-4 Hz. Fotosensibilità in circa il 20% dei casi Sensibilità alla deprivazione di sonno Recidiva alla sospensione della terapia nel 90% dei casi. Farmacoresistenza 10% dei casi Risposta a: VPA, PB, LTG. Peggioramento da CBZ e PHT

22
EPILESSIA con GRANDE MALE al RISVEGLIO
Esordio: anni Crisi tonico-cloniche d’emblée al risveglio oppure in ore serali EEG: normale attività di fondo, complessi punta e polipunta-onda irregolari a 3,5-4 Hz. Spesso EEG normale in più controll Risposta a: VPA, PB, CBZ….

23
EPILESSIA con MIOCLONIE PALPEBRALI ed ASSENZE (sindrome di Jeavons)
Esordio: 4-8 anni Prevalenza del sesso femminile Mioclonie rapide delle palpebre, talora associate a brevi mioclonie in retroversione del collo e a breve rottura del contatto GTC solo in età più tardiva, sporadici Scatenamento da parte delle variazioni di luce e dalla chiusura occhi EEG: polipunte e punta-onda indotti dalla chiusura degli occhi Risposta clinica incompleta

24
SINDROME DI WEST Può essere criptogenetica o sintomatica (tipico: sclerosi tuberosa) Esordio: 4-7 mesi Spasmi in flessione o in estensione Arresto dello sviluppo psicomotorio Risposta alla terapia spesso scarsa, con prognosi severa EEG: ipsaritmia Risposta a: ACTH, VGB

25
SINDROME DI LENNOX-GASTAUT
Può essere criptogenetica o sintomatica Esordio: 1-8 anni Associazione di: crisi atoniche, crisi toniche nel sonno. Assenze atipiche, crisi tonico-cloniche Arresto dello sviluppo psicomotorio Risposta alla terapia spesso scarsa, con prognosi severa EEG: punta-onda diffuse a 2 Hz o meno

26
EPILESSIA MIOCLONICA SEVERA dell’INFANZIA (sindrome di Dravet)
Esordio: 4-8 mesi con crisi febbrili Familiarità per epilessia generalizzata e CF Comparsa successiva di crisi non febbrili polimorfe Crisi cloniche prolungate Crisi miocloniche GTC Crisi parziali motorie con generalizzazione secondaria Regressione delle funzioni cognitive Farmacoresistenza quasi completa Peggioramento indotto da LTG (stato di male mioclonico)
")
27
EPILESSIE MIOCLONICHE PROGRESSIVE
Patologie geneticamente determinate caratterizzate da epilessia mioclonica, ma con sviluppo, più o meno precoce, di altri deficit neurologici (atassia, ritardo mentale, paresi, cecità…) Sviluppo progressivo di farmacoresistenza Sono esempi: sindrome di Unverricht-Lundborg, sindrome di Lafora, MERFF…
 Sviluppo progressivo di farmacoresistenza. Sono esempi: sindrome di Unverricht-Lundborg, sindrome di Lafora, MERFF…")
28
ALTRE EPILESSIE GENERAIZZATE IDIOPATICHE (Non classificate nel 1989, ma delineate come sindromi a sé stanti o classificate in altri gruppi) Epilessia con convulsioni febbrili plus Epilessia con mioclonie palpebrali ed assenze (sindrome di Jeavons) Epilessia mioclono-astatica Epilessia mioclonica severa dell’infanzia
 Epilessia mioclono-astatica. Epilessia mioclonica severa dell’infanzia.")
29
EPILESSIE PARZIALI IDIOPATICHE
EPILESSIA RIFLESSA DA LETTURA EPILESSIA A PAROSSISMI ROLANDICI E’ la forma più frequente di epilessia infantile Esordio: 3-9 anni Guarigione nel 100% dei casi Possibile non trattarle EPILESSIA IDIOPATICA OCCIPITALE Esordio 2-8 anni Sintomi visivi e/o vegetativi Prognosi buona EPILESSIA FRONTALE NOTTURNA FAMILIARE AUTOSOMICA DOMINANTE EPILESSIA TEMPORALE FAMILIARE

30
EPILESSIE PARZIALI SINTOMATICHE E CRIPTOGENETICHE
= EPILESSIE LOBARI EPILESSIA (-E) DEL LOBO TEMPORALE MESIALE e LATERALE la più frequente delle e. parziali; la più comune sindrome epilettica nell’adulto; spesso farmacoresistente esordio: bambino, giovane adulto anamnesi spesso positiva per: - familiarità - convulsioni febbrili
 DEL LOBO TEMPORALE. MESIALE e LATERALE. la più frequente delle e. parziali; la più comune sindrome epilettica nell’adulto; spesso farmacoresistente. esordio: bambino, giovane adulto. anamnesi spesso positiva per: - familiarità. - convulsioni febbrili.")
31
EPILESSIE PARZIALI TEMPORALI
CRISI PARZIALI SEMPLICI sintomi psichici (paura, angoscia) allucinazioni mnesiche, situazionali (déjà/jamais vu/veçu) fenomeni sensoriali: olfattivi, uditivi “aura” epigastrica CRISI PARZIALI COMPLESSE interruzione del contatto con l’ambiente staring (arresto motorio) automatismi, specie oro-alimentari (masticazione), ma anche gestuali segni vegetative (pallore, flushing, midriasi, orripilazione) durata superiore a 1’
 allucinazioni mnesiche, situazionali (déjà/jamais vu/veçu) fenomeni sensoriali: olfattivi, uditivi. aura epigastrica. CRISI PARZIALI COMPLESSE. interruzione del contatto con l’ambiente. staring (arresto motorio) automatismi, specie oro-alimentari (masticazione), ma anche gestuali. segni vegetative (pallore, flushing, midriasi, orripilazione) durata superiore a 1’")
32
EPILESSIE PARZIALI TEMPORALI
CRISI SECONDARIAMENTE GENERALIZZATA Confusione post-critica EEG intercritico: frequenti i falsi negativi anomalie “epilettiformi” (punte, sharp waves, complessi punta-onda) o onde lente uni- o bi-laterali
 o onde lente uni- o bi-laterali.")
33
EPILESSIE PARZIALI TEMPORALI
MESIALE paura ed altri sintomi emozionali allucinazioni olfattive sensazioni vegetative (epigastriche) LATERALE allucinazioni semplici, uditive, vestibolari o gustative afasia sensoriale fenomeni sensori-motori focali
 LATERALE. allucinazioni semplici, uditive, vestibolari o gustative. afasia sensoriale. fenomeni sensori-motori focali.")
34
EPILESSIE PARZIALI TEMPORALI
LOCALIZZAZIONE DEL FOCUS EPILETTOGENO: ipertono, se presente, è solitamente controlaterale automatismi spesso prevalenti ipsilateralmente Emisfero DOMINANTE afasia critica e post-critica maggiore durata dello stato post-critico (generalizzazione secondaria più frequente) (deviazione forzata controlaterale del capo) Emisfero NON DOMINANTE déjà vu verbalizzazione ictale allucinazioni visive complesse
 (deviazione forzata controlaterale del capo) Emisfero NON DOMINANTE. déjà vu. verbalizzazione ictale. allucinazioni visive complesse.")
35
EPILESSIE DEL LOBO FRONTALE
AREA SUPPLEMENTARE MOTORIA crisi posturali (schermidore), toniche focali CINGOLO crisi parziali complesse, automatismi gestuali motori complessi, sintomi autonomici, sintomi affettivi REGIONE ANTERIORE FRONTOPOLARE ORBITO-FRONTALE DORSOLATERALE OPERCOLARE masticazione, salivazione, deglutizione ... CORTECCIA MOTORIA crisi parziali semplici motorie, marcia jacksoniana, paralisi di Todd
, toniche focali. CINGOLO. crisi parziali complesse, automatismi gestuali motori complessi, sintomi autonomici, sintomi affettivi. REGIONE ANTERIORE FRONTOPOLARE. ORBITO-FRONTALE. DORSOLATERALE. OPERCOLARE. masticazione, salivazione, deglutizione ... CORTECCIA MOTORIA. crisi parziali semplici motorie, marcia jacksoniana, paralisi di Todd.")
36
CRISI PARZIALI COMPLESSE TEMPORALI FRONTALI
AURA: frequente, polimorfa DURATA: minuti Frequenza: plurimensile ESORDIO: staring AUTOMATISMI: semplici, oroalimentari VOCALIZZAZIONE: semplice POST-CRITICO: minuti AURA: più rara, poco specifica DURATA: secondi Crisi plurigiornaliere, spesso in cluster Frequenti crisi notturne vocalizzazione, espress.paura complessi e bizzarri bizzarra: urla, imprecazioni minimo o assente

37
CONVULSIONI FEBBRILI Il più comune disturbo critico dell’infanzia
(2 - 5 % di tutti i bambini di età inferiore a 5 anni) Crisi per lo più benigne Fattori genetici, età dipendenti (6 mesi - 2 anni) Recidiva nel 40% dei casi Solo il 2-4 % dei casi sviluppano in seguito epilessia (rischio maggiore per crisi prolungate e complesse) GEFS+
 Crisi per lo più benigne. Fattori genetici, età dipendenti (6 mesi - 2 anni) Recidiva nel 40% dei casi. Solo il 2-4 % dei casi sviluppano in seguito epilessia. (rischio maggiore per crisi prolungate e complesse) GEFS+")
38
EZIOLOGIA DELLE EPILESSIE
GENETICA: ipotizzata per le forme idiopatiche, verificata solo in alcune sindromi particolari MULTIFATTORIALE MENDELIANA (DOMINANTE, RECESSIVA, LEGATA A X, MITOCONDRIALE) ANOMALIE CROMOSOMICHE Monosomia 4p-, X fragile, sindrome di Angelman, trisomia 12p, cromosoma 20 ad anello, Down, Klinefelter
 ANOMALIE CROMOSOMICHE. Monosomia 4p-, X fragile, sindrome di Angelman, trisomia 12p, cromosoma 20 ad anello, Down, Klinefelter.")
39
EZIOLOGIA DELLE EPILESSIE
ANOMALIE DI SVILUPPO CORTICALE Lissencefalia Eterotopia a banda Eterotopia periventricolare Emimegalencefalia Schizencefalia Displasia corticale focale Sindrome di Aicardi LESIONI PRE- E PERINATALI MALATTIE INFETTIVE DEL SNC

40
EZIOLOGIA DELLE EPILESSIE
TUMORI CEREBRALI (10-15% delle epilessie dell’adulto) SCLEROSI MESIALE MALFORMAZIONI VASCOLARI MALATTIE CEREBRO-VASCOLARI TRAUMI CRANICI PATOLOGIE DEGENERATIVE SNC (m. di Alzheimer..)
 SCLEROSI MESIALE. MALFORMAZIONI VASCOLARI. MALATTIE CEREBRO-VASCOLARI. TRAUMI CRANICI. PATOLOGIE DEGENERATIVE SNC (m. di Alzheimer..)")
41
ANAMNESI EEG (intercritico, critico, Holter, video-EEG)
SINDROMICA DIAGNOSI EPILESSIA TC RM SPECT-PET TEST GENETICI EZIOLOGICA

42
EEG Consiste nella registrazione, dalla superficie dello scalpo, dell’attività elettrica spontanea della corteccia cerebrale Gli elettrodi sono disposti sullo scalpo sulla base di alcuni punti di repere (nasion ed inion) in modo che corrispondano alle aree corticali sottostanti. Il sistema di piazzamento e denominazione degli elettrodi è denominato “SISTEMA 10-20”.
 in modo che corrispondano alle aree corticali sottostanti. Il sistema di piazzamento e denominazione degli elettrodi è denominato SISTEMA")
43
EEG EEG: dati in favore di epilessia diagnosi sindromica
solo il 35% dei soggetti epilettici ha anomalie specifiche alla 1.a registrazione; un altro 50% presenta anomalie in registrazioni successive; il restante 15% non presenta mai anomalie

46
EEG EEG: metodiche di attivazione iperventilazione
stimolazione luminosa intermittente sonno Video-EEG “Holter”-EEG: registrazione prolungata in soggetti fuori dal laboratorio; utile per diagnosi differenziale Stereo-EEG: registrazione con elettrodi intracerebrali; solo in valutazione prechirurgica

47
EEG intercritico nell’epilessia: utilità
1. Diagnosi di positività 2. Diagnosi sindromica 3. Valutazione di improvviso deterioramento cognitivo 4. Indicazione chirurgica 5. Rischio di recidiva dopo la prima crisi 6. Rischio di recidiva dopo sospensione terapia NO (solitamente): valutazione del follow-up
: valutazione del follow-up.")
48
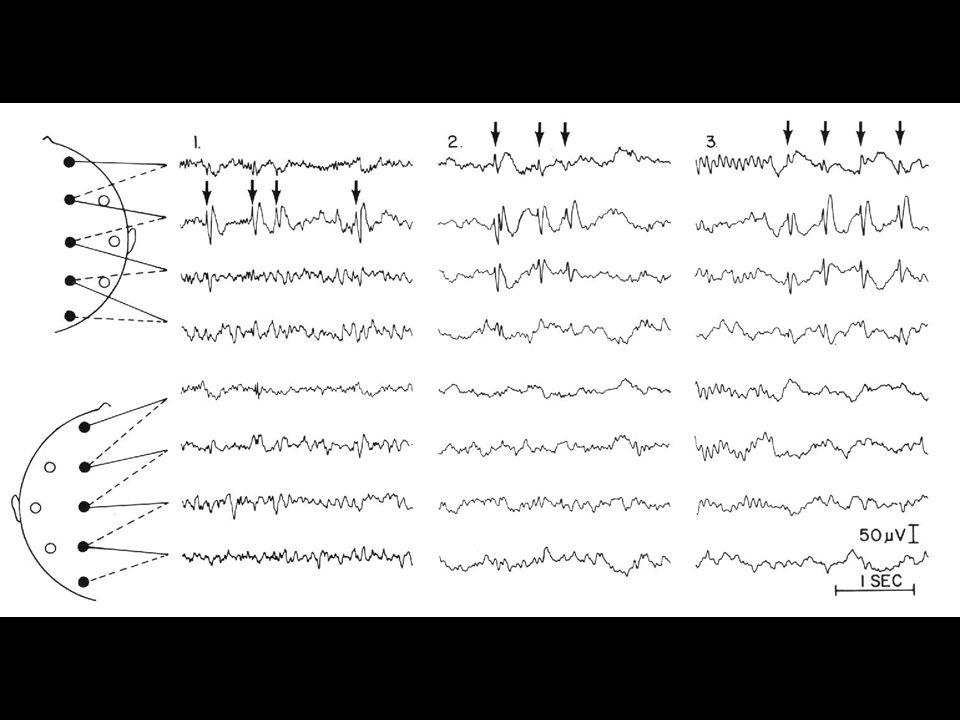
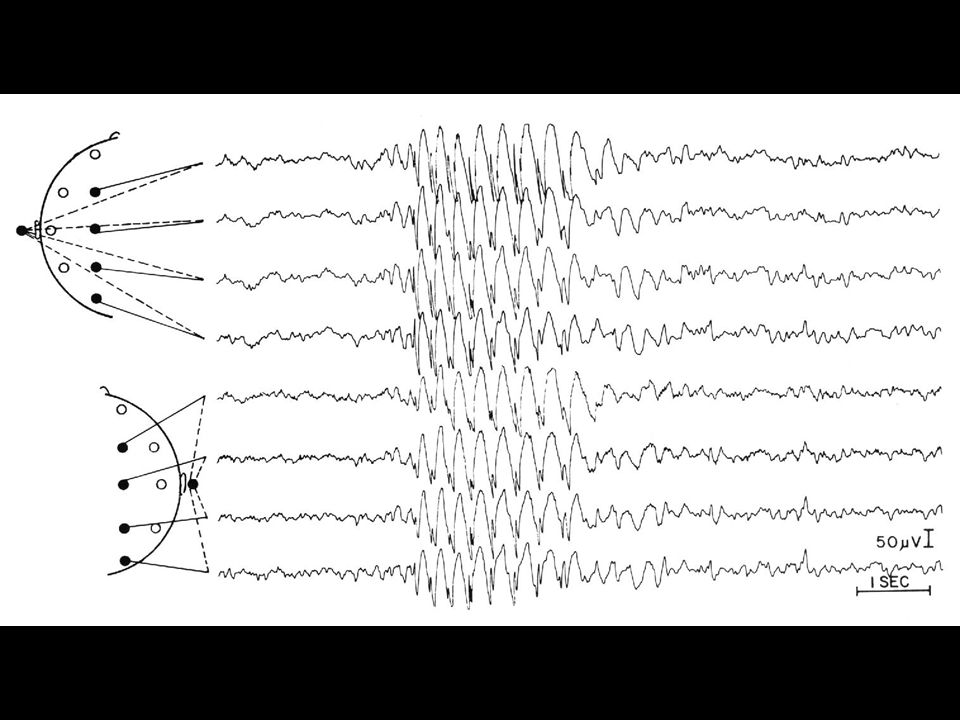
EEG: grafoelementi epilettiformi intercritici
1. Parossistici = chiaramente distinti dall’attività di fondo 2. Brusco cambiamento di polarità = morfologia puntuta 3. Durata inferiore a 200 ms 4. Corrispondenti ad un campo elettrico “fisiologico” inoltre, solitamente: 5. Polarità negativa 6. Seguiti da onda lenta

49
MONITORAGGIO VIDEO-EEG PROLUNGATO: Indicazioni (AEEG society, 1994)
DIAGNOSI… Di episodi critici di natura incerta Di episodi critici in pz epilettici, in caso di modificazioni sostanziali della semeiologia critica. CLASSIFICAZIONE… Della semeiologia critica in diagnosi sindromiche non precisate Del tipo e della frequenza delle anomalie critiche e intercritiche Della relazione degli episodi critici con stimoli specifici QUANTIFICAZIONE… Della frequenza di episodi critici in pz non in grado di segnalare o ricordare le crisi

50
TOMOGRAFIA COMPUTERIZZATA
Tecnica a raggi X Offre informazioni strutturali Vantaggi: poco costosa, rapida esecuzione Svantaggi: potere di risoluzione nettamente inferiore rispetto alla Rm

51
RISONANZA MAGNETICA Indagine strutturale
L’immagine viene costruita sulla base delle onde radio emesse dagli atomi di idrogeno stimolati da un campo magnetico Alta capacità di risoluzione Possibilità di ricostruzione dell’immagine in tre dimensioni

52
SCLEROSI TEMPORALE MESIALE

53
SCLEROSI TUBEROSA

54
GANGLIOGLIOMA

55
Epilessia: diagnosi differenziale
Lipotimia - Sincope Ipoglicemia Emicrania con aura Parasonnie e disturbi del sonno Attacchi ischemici transitori Disturbi del movimento (discinesia, distonie, tics) Patologie psichiatriche (attacchi di panico, crisi “isteriche” - “pseudocrisi”, simulazione, stati di depersonalizzazione, agitazione e automatismi in psicosi e oligofrenie)
 Patologie psichiatriche (attacchi di panico, crisi isteriche - pseudocrisi , simulazione, stati di depersonalizzazione, agitazione e automatismi in psicosi e oligofrenie)")
56
STORIA CLINICA DELLE EPILESSIE
Rischio di ricorrenza dopo prima crisi spontanea: 80% a 3 anni Risposta ottimale al trattamento: 70% Recidive dopo sospensione trattamento: dipendente dalla sindrome (20-50%) Farmacoresistenza: 20-30%
 Farmacoresistenza: 20-30%")
57
Epilessia: terapia Il trattamento viene solitamente iniziato dopo la 2.a crisi. Trattamento iniziale: monoterapia. Scelta del farmaco e posologia: individualizzate, in base a: tipo di crisi e sindrome età e peso tolleranza individuale al farmaco compliance coesistenza di altre malattie e altre terapie

58
Epilessia: terapia Fallimento 1.a monoterapia:
seconda monoterapia politerapia, sulla base delle possibili interazioni farmacocinetiche (minime) e farmacodinamiche (massime) Unico criterio di efficacia: andamento della frequenza critica. Il “dosaggio” plasmatico ha valore solo indicativo La persistenza di anomalie EEG non indica fallimento terapeutico
 e farmacodinamiche (massime) Unico criterio di efficacia: andamento della frequenza critica. Il dosaggio plasmatico ha valore solo indicativo. La persistenza di anomalie EEG non indica fallimento terapeutico.")
59
Epilessia: terapia, inizio
Rischio di epilessia no terapia Crisi unica no terapia Eccezioni: disturbo cerebrale progressivo EEG marcatamente alterato Due o più crisi monoterapia Eccezioni: crisi molto distanziate (> 1 anno) = oligoepilessia fattori precipitanti identificati (e. riflessa, farmaci, alccol, sostanze) probabile cattiva compliance (es. disturbo personalità) scelta del paziente o dei parenti
 = oligoepilessia. fattori precipitanti identificati (e. riflessa, farmaci, alccol, sostanze) probabile cattiva compliance (es. disturbo personalità) scelta del paziente o dei parenti.")
60
Epilessia: terapia, sospensione
Requisiti: assenza di ogni tipo di crisi da almeno 2-3 anni consenso informato del paziente Fattori favorevoli sfavorevoli epilessia infantile - esordio tardivo epilessia idiopatica - parziali assenza lesioni, ENO negativo breve durata malattia EEG normale non guidatore

61
Epilessia: farmaci antiepilettici (AED)
Meccanismi d’azione aumento inbizione GABA-mediata riduzione fenomeni eccitatori sinaptici (glutammato) riduzione delle scariche indotte da correnti Na+ e Ca++ voltaggio-dipendenti
 riduzione delle scariche indotte da correnti Na+ e Ca++ voltaggio-dipendenti.")
62
CARBAMAZEPINA (Tegretol ®)
Farmaco appartenente alla categoria dei triciclici, prime sperimentazioni negli anni’50 Agisce sulla conduttanza dei canali del Na+, riducendo il firing ripetitivo di potenziali di azione ad alta frequenza Proposti anche altri meccanismi, meno sicuri

63
CARBAMAZEPINA (Tegretol ®)
Esiste solo in formulazione orale, biodisponibilità 75-85%, con marcate variazioni interindividuali Metabolita attivo: 10,11 epossido Altamente metabolizzata a livello epatico Effetto induttore ed autoinduttore Molte interazioni con altri farmaci Principali effetti collaterali (30-50% dei pazienti) (necessaria sospensione in 5-10%): Sedazione, atassia, vertigine Nistagmo, diplopia Nausea, tremore, mioclonie Effetti idiosincrasici: Reazioni cutanee Agranulocitosi Epatopatia
 (necessaria sospensione in 5-10%): Sedazione, atassia, vertigine. Nistagmo, diplopia. Nausea, tremore, mioclonie. Effetti idiosincrasici: Reazioni cutanee. Agranulocitosi. Epatopatia.")
64
VALPROATO (Depakin ®) Sintetizzato come solvente nel 1882, utilizzato come AED dagli anni’60 Meccanismo di azione non ancora del tutto precisato Buone caratteristiche farmacocinetiche (con l’eccezione di elevato legame alle proteine) Disponibile anche in formulazione EV e retard (Depakin Chrono) Non induttore epatico, ma aumenta la concentrazione di PB e LTG
 Disponibile anche in formulazione EV e retard (Depakin Chrono) Non induttore epatico, ma aumenta la concentrazione di PB e LTG.")
65
VALPROATO (Depakin ®) Buona la tolleranza, ma effetti collaterali di tipo endocrino, specie nelle donne, e alcuni effetti idiosincrasici gravi (pancreatiti, epatiti fulminanti anche fatali, specie nei bambini < 2 anni) Teratogeno più di altri farmaci Efficacia marcata, ad ampio spettro. Viene considerato farmaco di prima linea in pressochè tutti i tipi di crisi
 Teratogeno più di altri farmaci. Efficacia marcata, ad ampio spettro. Viene considerato farmaco di prima linea in pressochè tutti i tipi di crisi.")
66
Stato di male epilettico
Crisi ripetute e prolungate due o più crisi senza completo ricupero delle funzioni neurologiche tra le crisi attività critica più o meno continua per 30’ o più Classificazione: generalizzato convulsivo parziale semplice (epilessia parziale continua) non convulsivo parziale complesso assenza
 non convulsivo. parziale complesso. assenza.")
67
Stato di male epilettico: terapia
Accesso venoso; urgenze ematologiche; tiamina 100 mg; glucosata 50% 50 cc Lorazepam i.v. (0,1 mg/kg [< 2 mg/min]), oppure diazepam (0,2 mg/kg [< 5 mg/min]) Se inefficace: fenitoina i.v. (Aurantin) 20 mg(kg < 50 mg/min) monitorando P.A. ed ECG id.: ulteriori 20 mg(kg fenitoina i.v. id.: assistenza respiratoria; fenobarbitale 20 mg/kg id.: coma barbiturico (pentobarbital) a dosi sopprimenti attività EEG
, oppure diazepam (0,2 mg/kg [< 5 mg/min]) Se inefficace: fenitoina i.v. (Aurantin) 20 mg(kg < 50 mg/min) monitorando P.A. ed ECG. id.: ulteriori 20 mg(kg fenitoina i.v. id.: assistenza respiratoria; fenobarbitale 20 mg/kg. id.: coma barbiturico (pentobarbital) a dosi sopprimenti attività EEG.")
68
Epilessia: terapia chirurgica
Riservata alle epilessie farmacoresistenti gravi, con importanti limitazioni delle vita quotidiana Interventi palliativi callosotomia, transezioni subpiali, emisferectomia cercano di migliorare la frequenza critica, senza pretese di guarigione Interventi curativi asportazione del focolaio epilettogeno possibili solo in epilessie a focolaio unico, localizzato in sedi la cui asportazione non comporti deficit neurologici valutazione prechirurgica approfondita

Presentazioni simili
>")
, sta ad indicare una modalità di reazione.>")









>")



 da fattori potenzialmente dannosi per le cellule. Le cellule “offese” liberano molecole che.>")



