Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Ruolo del trasporto assonale nelle malattie neurodegenerative
Molte delle grandi malattie neurodegenerative umane mostrano patologia assonale. Tali patologie evidenziano danni all’assone come parte del processo patogeno e, in particolare, danni al trasporto di carichi attraverso gli assoni. Infatti, ora sappiamo che l'alterazione del trasporto assonale è un evento precoce e forse causale in molte di queste malattie. Qui, passiamo in rassegna il ruolo del trasporto assonale nella malattia neurodegenerativa.

2
I neuroni hanno un corpo cellulare che si prolunga con assoni e dendriti.

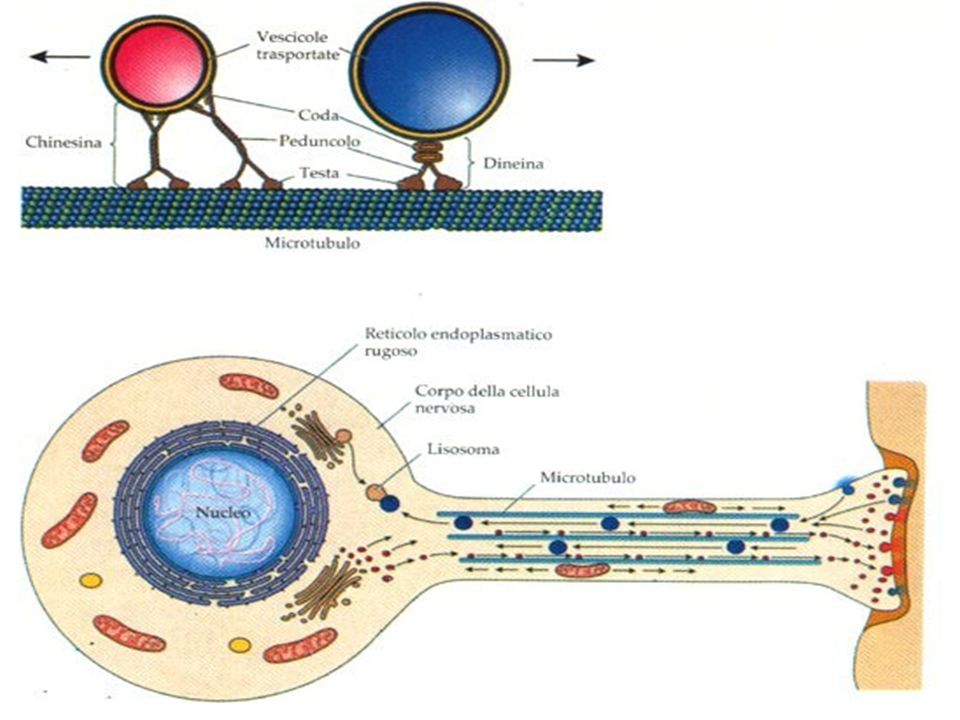
3
Trasporto assonale Il trasporto intracellulare di proteine e organelli carichi è un requisito essenziale per tutte le cellule, soprattutto per i neuroni. Il trasporto assonale può essere: “lento”: muove materiale dal soma al terminale assonale, trasporta componenti che non sono consumati rapidamente dalla cellula, come enzimi e proteine del citoscheletro; “veloce”: è bidirezionale; il trasporto anterogrado (in avanti) trasporta vescicole sinaptiche e secretorie dal soma al terminale assonale, mentre il trasporto retrogrado (all’indietro) trasporta vecchie componenti di membrana dal terminale assonale al soma perché siano riciclate. La dineina interviene nel trasporto anterogrado e la chinesina in quello retrogrado. Le due proteine motrici dineina e chinesina, impegnate nel trasporto di vescicole, idrolizzando ATP, si spostano lungo il microtubulo grazie a cambiamenti di conformazione dei peduncoli che le legano ai microtubuli.
 trasporta vescicole sinaptiche e secretorie dal soma al terminale assonale, mentre il trasporto retrogrado (all’indietro) trasporta vecchie componenti di membrana dal terminale assonale al soma perché siano riciclate. La dineina interviene nel trasporto anterogrado e la chinesina in quello retrogrado. Le due proteine motrici dineina e chinesina, impegnate nel trasporto di vescicole, idrolizzando ATP, si spostano lungo il microtubulo grazie a cambiamenti di conformazione dei peduncoli che le legano ai microtubuli.")
5
Motori molecolari I motori molecolari generano forza dall’idrolisi dell'ATP per spostare carichi lungo le piste del citoscheletro. I principali motori che si muovono sui microtubuli sono membri della superfamiglia della chinesina e della dineina citoplasmatica. La chinesina-1 è il motore molecolare più studiato ed è un eterotetramero di due catene pesanti KHC e due catene leggere KLC. KHC è composto da un dominio catalitico motore, una breve regione linker del collo, α-elica a spirale, che viene interrotto da due regioni di cerniera, e la coda. Il dominio motore si lega ai microtubuli, contiene l'ATPasi, e, insieme al collo, conferisce direzionalità; la coda, invece, unitamente a KLC, lega il carico e regola l'attività motoria. La dineina citoplasmatica è un complesso multisubunità che contiene due catene pesanti che sono associate, a loro volta, con catene intermedie e catene leggere. Le catene pesanti portano l’ATPasi e legano i microtubuli, mentre le altre catene sono coinvolti nel legame cargo e vincolante per la dinactina. La dinactina è un complesso proteico che contiene p150glued e dynamitin.

6
Motori molecolari Tail Stalk Motor domain Dineina Chinesina 1
Dynamitin Dynactin Tail Kinesin light chain (KLC) Intermediate chain Heavy chain Stalk p150glued Kinesin heavy chain (KHC) Motor domain Motor domain Dineina Chinesina 1
 Intermediate chain. Heavy chain. Stalk. p150glued. Kinesin heavy chain. (KHC) Motor domain. Motor domain. Dineina. Chinesina 1.")
7
I meccanismi attraverso i quali il trasporto assonale è interrotto sono molteplici.
Come un viaggio in treno, il disagio per il trasporto può avvenire tramite: danni ai motori (chinesina e dineina citoplasmatica); danni alle rotaie (microtubuli); danni ai carichi (ad esempio, per inibire il loro attaccamento ai motori); danni alla fornitura di combustibile (ATP). Tutti questi insulti possono contribuire alla neurodegenerazione.
; 2. danni alle rotaie (microtubuli); 3. danni ai carichi (ad esempio, per inibire il loro attaccamento ai motori); danni alla fornitura di combustibile (ATP). Tutti questi insulti possono contribuire alla neurodegenerazione.")
8
Il trasporto assonale nelle malattie neurodegenerative
Il trasporto assonale difettoso delle cellule del corpo crea accumuli di organelli e altre proteine, che sono tratti distintivi di molte malattie neurodegenerative umane. Insieme, tali patologie suggeriscono che il funzionamento difettoso dell’assone contribuisce alla malattia e, in particolare, che il danno al trasporto assonale può essere alla base dell’accumulo patogenetico di organelli.

9
MALATTIA DI ALZHEIMER E DISTURBI CORRELATI
Due delle caratteristiche patologiche dell'Alzheimer sono i grovigli neurofibrillari, contenenti filamenti appaiati elicoidali (PHFs) e le placche amiloidi. PHFs sono assemblati da Tau iperfosforilate (una proteina assonica associata ai microtubuli), mentre, le placche amiloidi sono formazioni extracellulari costituite da una parte centrale in cui si accumula proteina amiloide, e una parte periferica in cui si depositano detriti neuronali. La proteina amiloide (A-beta) è il maggior costituente delle placche amiloidi, ed ha origine dalla proteina APP.
 e le placche amiloidi. PHFs sono assemblati da Tau iperfosforilate (una proteina assonica associata ai microtubuli), mentre, le placche amiloidi sono formazioni extracellulari costituite da una parte centrale in cui si accumula proteina amiloide, e una parte periferica in cui si depositano detriti neuronali. La proteina amiloide (A-beta) è il maggior costituente delle placche amiloidi, ed ha origine dalla proteina APP.")
10
Dall'analisi post-mortem di tessuti cerebrali di pazienti affetti da Alzheimer, si è potuto riscontrare un accumulo extracellulare di questa proteina, chiamata, per l’appunto, APP. La APP (Proteina Progenitrice dell'Amiloide) che viene prodotta, è degradata durante un processo che vede coinvolti tre enzimi che operano tagli proteolitici: la α-secretasi e la β-secretasi in un primo momento e successivamente la γ-secretasi. Attraverso due tagli successivi operati prima dall'α-secretasi e poi dall'γ-secretasi, viene prodotto un peptide innocuo chiamato p3. La β-secretasi, invece, opera un taglio differente che porta alla produzione di due peptidi di 40 e 42 aminoacidi, chiamati beta-amiloide (Aβ 1-40 e Aβ 1-42): il secondo (Aβ 1-42) è considerato il più tossico a livello neuronale. Nei soggetti sani il processo di degradazione della APP sembra essere operato principalmente dalla α-secretasi. Per motivi non totalmente chiariti, nei soggetti malati l'enzima che interviene sull'APP non è l'α-secretasi ma la β-secretasi, con una larga produzione di proteina beta-amiloide. Tale beta-amiloide non presenta le caratteristiche biologiche della forma naturale, ma tende a depositarsi in aggregati extracellulari sulla membrana dei neuroni.
 che viene prodotta, è degradata durante un processo che vede coinvolti tre enzimi che operano tagli proteolitici: la α-secretasi e la β-secretasi in un primo momento e successivamente la γ-secretasi. Attraverso due tagli successivi operati prima dall α-secretasi e poi dall γ-secretasi, viene prodotto un peptide innocuo chiamato p3. La β-secretasi, invece, opera un taglio differente che porta alla produzione di due peptidi di 40 e 42 aminoacidi, chiamati beta-amiloide (Aβ 1-40 e Aβ 1-42): il secondo (Aβ 1-42) è considerato il più tossico a livello neuronale. Nei soggetti sani il processo di degradazione della APP sembra essere operato principalmente dalla α-secretasi. Per motivi non totalmente chiariti, nei soggetti malati l enzima che interviene sull APP non è l α-secretasi ma la β-secretasi, con una larga produzione di proteina beta-amiloide. Tale beta-amiloide non presenta le caratteristiche biologiche della forma naturale, ma tende a depositarsi in aggregati extracellulari sulla membrana dei neuroni.")
11
Le mutazioni e le duplicazioni del gene che codifica per la APP e le mutazioni del gene che codifica per la presenilina, causano alcune forme familiari della malattia di Alzheimer. La fosforilazione di KLC causa una mutazione del gene presenilina da GSK3β che inibisce la chinesina, interrompendo l’attaccamento della chinesina alle vescicole. Mutant presenilin GSK3β

12
La fosforilazione di KHC ad opera dell’enzima c-Jun-N-terminal kinase (JNK), inibisce la chinesina ostacolando le interazioni di KHC con microtubuli. JNK

13
Ulteriori studi mettono in evidenza che nei malati di Alzheimer interviene un ulteriore meccanismo patologico: all'interno dei neuroni una Proteina-Tau, fosforilata in maniera anomala, si accumula nei cosiddetti "aggregati neurofibrillari" (o ammassi neurofibrillari). Una possibilità è che Tau interferisca con il legame della chinesina al microtubulo causando danni al trasporto assonale.

14
Danni ai microtubuli Ulteriori difetti dei meccanismi di trasporto assonale possono essere dovuti a danni ai microtubuli, strutture altamente dinamiche che subiscono periodi di rapida crescita e contrazione, e il loro comportamento dinamico è regolato da diversi meccanismi. La deregolamentazione di queste proprietà dinamiche può portare alla rottura delle componenti cellulari implicate nel trasporto assonale.

15
L’iperfosforilazione della proteina Tau può portare ad una destabilizzazzione del trasporto assonale dovuto principalmente alla distruzione dei microtubuli.

16
Perdita di funzione dettata dalla mutazione delle proteine spastina/atlastina, che inducono raggruppamento anomalo di microtubuli alterando il trasporto.

17
Mutante SOD1. Le mutazioni del gene che codifica Cu / Zn superossido dismutasi -1 (SOD1) causano il 20% dei casi familiari di SLA. L'espressione di SOD1 mutata in topi transgenici induce la malattia del motoneurone (MND), e l'analisi di tali topi rivelano che il danno al trasporto assonale è un evento patogeno precoce. La natura precoce di questo danno dimostra che i danni al trasporto assonale contribuiscono allo sviluppo della malattia già dalle fasi iniziali, e non solo nello stadio terminale. Le mutazioni di SOD1 causano danni sia al trasporto assonale veloce che a quello lento. Tuttavia, le mutazioni di SOD1 colpiscono in maniera differente il trasporto assonale di carichi specifici. Il movimento anterogrado delle componenti del citoscheletro è rallentato, il trasporto veloce delle vescicole è inibito in entrambe le direzioni, anterograda e retrograda, ma l'inibizione del movimento mitocondriale è anterograda specifica.
, e l analisi di tali topi rivelano che il danno al trasporto assonale è un evento patogeno precoce. La natura precoce di questo danno dimostra che i danni al trasporto assonale contribuiscono allo sviluppo della malattia già dalle fasi iniziali, e non solo nello stadio terminale. Le mutazioni di SOD1 causano danni sia al trasporto assonale veloce che a quello lento. Tuttavia, le mutazioni di SOD1 colpiscono in maniera differente il trasporto assonale di carichi specifici. Il movimento anterogrado delle componenti del citoscheletro è rallentato, il trasporto veloce delle vescicole è inibito in entrambe le direzioni, anterograda e retrograda, ma l inibizione del movimento mitocondriale è anterograda specifica.")
18
SOD-1 è un enzima con funzione antiossidante in quanto riduce il livello di ione superossido (O2−), un radicale libero tossico prodotto durante il metabolismo ossidativo cellulare capace di alterare le proteine, le membrane e il DNA stesso.
, un radicale libero tossico prodotto durante il metabolismo ossidativo cellulare capace di alterare le proteine, le membrane e il DNA stesso.")
19
Molti studi hanno dimostrato che la mutazione della SOD1 si associa selettivamente con il danno ai mitocondri. Questo danno è in grado di mettere in pericolo la catena mitocondriale di trasferimento degli elettroni e così la sintesi di ATP. L’inibizione del trasporto mitocondriale anterogrado porta ad un aumento netto del loro trasporto retrogrado, (che si traduce in una deplezione degli stessi mitocondri). Questo potrebbe influenzare negativamente il trasporto assonale di altri carichi a causa di diminuiti livelli di ATP.
. Questo potrebbe influenzare negativamente il trasporto assonale di altri carichi a causa di diminuiti livelli di ATP.")
20
Difetti nei meccanismi di trasporto assonale possono comportare gravi danni ai mitocondri, e infatti questi danni sono presenti in molte malattie neurodegenerative, e la disfunzione mitocondriale probabilmente influisce trasporto assonale in almeno due modi: l’inibizione delle funzioni mitocondriali riduce il trasporto anterogrado sia dei mitocondri che delle vescicole forse per mezzo dell’attivazione di PKCδ. La conseguente diminuzione dei mitocondri negli assoni farà probabilmente diminuire la fornitura di ATP ai motori molecolari portando alla diminuzione del movimento anterogrado e retrogrado di altri carichi assoplasmatici.

22
Poiché la disfunzione mitocondriale riduce anche la produzione di ATP, quest'ultima potrebbe essere parte di un circolo vizioso che conduce infine al «dying-back» degli assoni (morte retrograda).
.")
23
Anche se le mutazioni della SOD-1 possono essere neurone specifica, si è dimostrato che possono contribuire anche altri tipi di cellule allo sviluppo della malattia. Quindi, anche se l’espressione di SOD1 mutante nei neuroni motore è un fattore fondamentale per l’insorgenza della malattia, l’espressione nella microglia (Le cellule della microglia sono un tipo di cellule della glia che si occupano della prima e principale difesa immunitaria attiva nel sistema nervoso centrale) influenza notevolmente la progressione della malattia. Risposte infiammatorie sono state fortemente collegate alla SLA , e la proteina SOD1 mutante stessa può essere secreta e attivare la microglia. Segnali infiammatori dalle cellule vicine possono quindi fornire ulteriori insulti al trasporto assonale.
 influenza notevolmente la progressione della malattia. Risposte infiammatorie sono state fortemente collegate alla SLA , e la proteina SOD1 mutante stessa può essere secreta e attivare la microglia. Segnali infiammatori dalle cellule vicine possono quindi fornire ulteriori insulti al trasporto assonale.")
24
Insulti eccitotossici probabilmente contribuiscono alla SLA perché alterazioni di proteine e di metaboliti coinvolti nella gestione del glutammato, sono presenti in casi di SLA sporadici. Inoltre, la proteina SOD-1 mutante danneggia selettivamente il trasportatore gliale del glutammato EAAT2 (che rimuove il glutammato sinaptico) e i livelli di EAAT2 sono ridotti nei topi transgenici che presentano mutazione della SOD-1. In particolare, il glutammato attiva JNK, p38, e cdk5/p35 (tutti sono fortemente collegati al trasporto assonale).
 e i livelli di EAAT2 sono ridotti nei topi transgenici che presentano mutazione della SOD-1. In particolare, il glutammato attiva JNK, p38, e cdk5/p35 (tutti sono fortemente collegati al trasporto assonale).")
25
Così JNK e p38 fosforilano rispettivamente le catene pesanti e leggere della chinesina, per inibire il trasporto.

26
DANNI AI CARICHI Infine, la mutazione di SOD1 può danneggiare i carichi in modo da inibire il loro trasporto (forse promuovendo il loro rilascio dai motori). L’accumulo di neurofilamenti garantisce la presenza della SLA, mentre la sovrespressione delle catene leggere (NFL) e pesanti (NFH), la periferina (un’altra proteina neuronale del filamento intermedio), o un mutante NFL che disturbi l’assemblaggio del neurofilamento induce MND nei topi transgenici, quando invece sarebbe necessario il loro corretto montaggio perché possa avvenire il trasporto (in questi neurofilamenti transgenici infatti il trasporto assonale è difettoso).
. L’accumulo di neurofilamenti garantisce la presenza della SLA, mentre la sovrespressione delle catene leggere (NFL) e pesanti (NFH), la periferina (un’altra proteina neuronale del filamento intermedio), o un mutante NFL che disturbi l’assemblaggio del neurofilamento induce MND nei topi transgenici, quando invece sarebbe necessario il loro corretto montaggio perché possa avvenire il trasporto (in questi neurofilamenti transgenici infatti il trasporto assonale è difettoso).")
27
Tutto ciò è stato dimostrato grazie a esperimenti su topi transgenici con SOD-1 mutante sono stati incrociati con neurofilamenti transgenici, e modulando l’espressione di questi ultimi la malattia risulta sensibilmente alterata. Infatti la delezione dei domini laterali di NFM e NFH risulta fortemente protettiva nei confronti della malattia provocata dal mutante SOD-1.

28
MORBO DI HUNTINGTON Alcune malattie neurodegenerative familiari sono causate dalla dilatazione di tratti di poliglutammina (un frammento peptidico consistente in una ripetizione dell’aminoacido glutamina), e tali malattie comprendono la malattia di Huntington , malattia di Kennedy , e alcune atassie spino-cerebellari. Nella corea di Huntington e nella malattia di Kennedy, le espansioni avvengono all'interno delle proteine huntingtina e recettore androgeno, e l'espressione di huntingtina mutante o del recettore androgeno sconvolge il trasporto assonale in molti modelli , tra cui i neuroni dei mammiferi. La poliglutammina espansa nel recettore androgeno porta alla fosforilazione di KHC e questa forsforilazione fa ridurre il legame della chinesina ai microtubuli inibendo il trasporto.
, e tali malattie comprendono la malattia di Huntington , malattia di Kennedy , e alcune atassie spino-cerebellari. Nella corea di Huntington e nella malattia di Kennedy, le espansioni avvengono all interno delle proteine huntingtina e recettore androgeno, e l espressione di huntingtina mutante o del recettore androgeno sconvolge il trasporto assonale in molti modelli , tra cui i neuroni dei mammiferi. La poliglutammina espansa nel recettore androgeno porta alla fosforilazione di KHC e questa forsforilazione fa ridurre il legame della chinesina ai microtubuli inibendo il trasporto.")
29
Tuttavia nella corea di Huntington ci possono essere altri meccanismi che portano alla danneggiamento dei trasporti, e alcuni di questi meccanismi comportano la perdita di funzione dell’ huntingtina; infatti l’huntingtina lega HAP-1, che è fortemente implicata nei processi di trasporto, e inoltre migliora il trasporto retrogrado di BDNF. L’ huntingtina mutante è difettoso in questo processo. In conclusione le poliglutammine espanse possono quindi interferire con il trasporto assonale a causa della fosforilazione di KHC, e huntingtina mutante può danneggiare il trsporto influendo sia sui motori molecolari che sui microtubuli.

Presentazioni simili