Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
La nostra esperienza all’Istituto Ebri-CNR
Tipizzazione del gene PV 92 Per Alu

2
Individuazione di Elementi Alu sul cromosoma 16
Come è noto il nostro corredo cromosomico è costituito da un numero diploide pari a 46 cromosomi PV92 è un gene localizzato sul cromosoma 16 L’inserimento di un retrotrasposone Alu avvenne, come evento mutazionale, in un soggetto (fondatore) migliaia di anni fa I possibili genotipi del PV92 per ALU sono: +/+ +/- -/- Questa tipizzazione è usati in genetica delle popolazioni, in analisi di paternità e in medicina forense. Non si ha alcuna associazione con fenotipi.
 migliaia di anni fa. I possibili genotipi del PV92 per ALU sono: +/+ +/- -/- Questa tipizzazione è usati in genetica delle popolazioni, in analisi di paternità e in medicina forense. Non si ha alcuna associazione con fenotipi.")
3
Cromosoma 16 PV92 ALU Cromosoma 16 Alu PV92 Alu PV92

4
PCR “reazione a catena della polimerasi”
Nel 1989 è stato messo a punto un processo, noto come PCR (reazione a catena della polimerasi), in grado di sintetizzare milioni di copie di un segmento di DNA in tempi assai brevi e con una procedura relativamente semplice La PCR richiede una certa conoscenza del segmento da copiare ed amplificare, di cui deve essere nota una sequenza di circa due dozzine di basi, all’estremità 3’ dei due filamenti che costituiscono il segmento da amplificare Infatti è necessario predisporre due iniziatori specifici, di circa basi, che possano favorire l’innesco della duplicazione del frammento di DNA da amplificare, riconoscendone le estremità 3’ dalle quali inizia la duplicazione Grazie all’unicità delle sequenze del DNA , i due iniziatori (PRIMER) legheranno solo la regione del DNA per la quale sono stati predisposti. Quindi in una preparazione contenente l’intero genoma umano, costituito da tre miliardi di coppie di basi, gli iniziatori predisposti interagiranno solo con il segmento genico sul quale si sta lavorando.
, in grado di sintetizzare milioni di copie di un segmento di DNA in tempi assai brevi e con una procedura relativamente semplice. La PCR richiede una certa conoscenza del segmento da copiare ed amplificare, di cui deve essere nota una sequenza di circa due dozzine di basi, all’estremità 3’ dei due filamenti che costituiscono il segmento da amplificare. Infatti è necessario predisporre due iniziatori specifici, di circa basi, che possano favorire l’innesco della duplicazione del frammento di DNA da amplificare, riconoscendone le estremità 3’ dalle quali inizia la duplicazione. Grazie all’unicità delle sequenze del DNA , i due iniziatori (PRIMER) legheranno solo la regione del DNA per la quale sono stati predisposti. Quindi in una preparazione contenente l’intero genoma umano, costituito da tre miliardi di coppie di basi, gli iniziatori predisposti interagiranno solo con il segmento genico sul quale si sta lavorando.")
5
3) Valutazione dei risultati
Regione da amplificare PV92 Senza Alu 415 bp Regione da amplificare PV92 Con Alu 715bp 300bp

6
I banconi sono organizzati con micropipette e relativi puntali, provette e portaprovette, kit con reagenti, in postazioni predisposte per due allievi che lavoreranno individualmente. Tutto il materiale viene siglato da ogni allievo con un numero che consenta, in forma anonima,l’attribuzione della tipizzazione.

7
1. Viene effettuato uno sciacquo boccale energico con soluzione fisiologica, per 1 minuto. Lo sciacquo viene raccolto nel bicchiere usato da ogni allievo.

8
2. Viene prelevato con opportuna micropipetta 1 ml dello sciacquo boccale che viene deposto in una provetta da centrifuga. Per ogni micropipetta si utilizza il corrispondente puntale usa e getta.

9
3. Si effettua una centrifugazione di 1 minuto a 14.000 rpm.

10
4. Si verifica la formazione, per centrifugazione, di un sedimento sufficiente (pellett di cellule) e si svuota la provetta eliminando il sovranatante. Rimane una piccola quantità di sospensione contenente le CELLULE della MUCOSA BUCCALE.
 e si svuota la provetta eliminando il sovranatante. Rimane una piccola quantità di sospensione contenente le CELLULE della MUCOSA BUCCALE..")
11
5. Si agita la provettina per sospendere adeguatamente le cellule, si prelevano 30 μl della sospensione che vengono depositati in una nuova provetta predisposta contenente: a) sostanze basiche necessarie per la solubilizzazione degli acidi grassi presenti nei fosfolipidi di membrana, in tal la cellula ed il nucleo si lisano; il principio è lo stesso dei saponi che usiamo per lavarci le mani b) resina che serve ad allontanare gli ioni metallici che successivamente inibirebbero la DNA polimerasi
 sostanze basiche necessarie per la solubilizzazione degli acidi grassi presenti nei fosfolipidi di membrana, in tal la cellula ed il nucleo si lisano; il principio è lo stesso dei saponi che usiamo per lavarci le mani. b) resina che serve ad allontanare gli ioni metallici che successivamente inibirebbero la DNA polimerasi.")
12
6. la provetta viene sottoposta a centrifugazione per 1 minuto a 14
6. la provetta viene sottoposta a centrifugazione per 1 minuto a rpm, poi posta per 10 minuti in bagnomaria a 100° C ed ancora centrifugata per 1 minuto a rpm. Si ottiene così un precipitato contenente il materiale cellulare ormai inutile mentre nel sovranatante rimane il DNA.

13
E al Socrate………DNA e KIWI
La prima parte dell’esperienza di tipizzazione genica, cioè l’estrazione deI DNA dalle cellule è stata sperimentata anche nel nostro laboratorio dagli allievi de quarto anno C. Ecco il protocollo dell’ esperienza: Materiali e Metodo: un kiwi abbastanza maturo, un sacchetto robusto di plastica, sale da cucina, sapone liquido, garza, alcool isopropilico (in alternativa si può usare l'alcool etilico), vetreria. Sbucciare il frutto e tagliarlo in piccoli pezzi da inserire nel sacchetto di plastica. Chiudere il sacchetto con un elastico oppure facendo un nodo sull'apertura, cercando di togliere prima tutta l'aria.Frantumare la polpa con le mani, per circa 2 minuti.
, vetreria. Sbucciare il frutto e tagliarlo in piccoli pezzi da inserire nel sacchetto di plastica. Chiudere il sacchetto con un elastico oppure facendo un nodo sull apertura, cercando di togliere prima tutta l aria.Frantumare la polpa con le mani, per circa 2 minuti.")
14
Intanto preparare una soluzione salina, mettendo un cucchiaino da tè di sale in 100 millilitri circa di acqua. Quando la polpa è stata ridotta in poltiglia, aggiungere 10 millilitri della soluzione salina, chiudere di nuovo il sacchetto e continuare a schiacciare la polpa per altri 5 minuti.Nel frattempo preparare una soluzione con il sapone liquido (3-4 cucchiaini di sapone in 30 millilitri circa di acqua, mescolare senza sbattere per evitare di produrrete troppa schiuma). Preparare anche la garza per la filtrazione, mettendone alcuni strati sull'imboccatura di un bicchiere e fermando il tutto con l'elastico. Filtrare la polpa.
. Preparare anche la garza per la filtrazione, mettendone alcuni strati sull imboccatura di un bicchiere e fermando il tutto con l elastico. Filtrare la polpa.")
15
Nel filtrato aggiungere 3 millilitri circa di sapone liquido diluito
Nel filtrato aggiungere 3 millilitri circa di sapone liquido diluito. Mescolare delicatamente per circa 1 minuto. Ora la fase forse più delicata: tenendo inclinato il contenitore con il filtrato, versare una quantità circa doppia, rispetto al filtrato, di alcool isopropilico, facendo attenzione a non mescolare i liquidi. Bisogna far formare due strati, dato che l'alcool è più leggero del miscuglio acquoso. Per ottenere migliori risultati è utile tenere l'alcool in frigo per 12 ore circa, prima di utilizzarlo. Più è freddo, meglio è. L'alcool rende il DNA insolubile e quindi visibile come una “massa” biancastra.

16
TORNIAMO ALLA TIPIZZAZIONE
7. Con la corrispondente micropipetta si prelevano con cura, evitando di sospendere nuovamente il precipitato, 30 μl del solo sovranatante (DNA) che si deposita in una nuova cuvetta che fornirà 2,5 μl di materiale con il quale continuare l’esperienza, mentre i 17,5 μl residui saranno accantonati per avere materiale di riserva in caso di errori. E’ un passaggio in più che viene seguito per cautela.
 che si deposita in una nuova cuvetta che fornirà 2,5 μl di materiale con il quale continuare l’esperienza, mentre i 17,5 μl residui saranno accantonati per avere materiale di riserva in caso di errori. E’ un passaggio in più che viene seguito per cautela.")
17
8. A questo punto si lavora con tre provettine:
la prima contenente 2,5 μl con il DNA ottenuto dalle cellule boccali,

18
la seconda, fornita dal kit, contenente un agente acido in grado di neutralizzare il precedente passaggio che ha reso basica la soluzione di DNA e il primer, cioè la breve sequenza di nucleotidi che deve riconoscere il punto di innesco della duplicazione del tratto di DNA da amplificare

19
la terza, fornita dal kit, contenete la TaQ polimerasi, cioè l’enzima termostabile capace di catalizzare la duplicazione del DNA anche ad elevate temperature, oltre a sali minerali con funzione tamponante e ai desossinucleotidi trifosfati necessari per la duplicazione della regione PV92

20
Si prelevano con opportune e distinte micropipette 2,5 μl dalla prima provetta e 22,5 μl dalla seconda inserendo quanto prelevato nella terza provetta contenente la TaQ polimerasi.

21
9. Ora si passa in PCR (REAZIONE A CATENA della POLIMERASI), tutte le cuvette predisposte da ogni allievo vengono inserite nel TERMICICLATORE che viene avviato per compiere 30 cicli di duplicazione del DNA per un totale di 1 ora e 15 minuti. Più precisamente il termociclatore viene regolato per alternare 20 secondi a 95°C ad 1 minuto a 60° C allo scopo di passare da una denaturazione del DNA (con apertura della doppia elica per effetto dell’elevata temperatura) ad una duplicazione (appaiamento ed estensione), catalizzata dalla TaQ polimerasi, dei tratti delle singole eliche innescati dai primer utilizzati.
, tutte le cuvette predisposte da ogni allievo vengono inserite nel TERMICICLATORE che viene avviato per compiere 30 cicli di duplicazione del DNA per un totale di 1 ora e 15 minuti. Più precisamente il termociclatore viene regolato per alternare 20 secondi a 95°C ad 1 minuto a 60° C allo scopo di passare da una denaturazione del DNA (con apertura della doppia elica per effetto dell’elevata temperatura) ad una duplicazione (appaiamento ed estensione), catalizzata dalla TaQ polimerasi, dei tratti delle singole eliche innescati dai primer utilizzati..")
22
I 30 cicli predisposti producono milioni di copie del tratto di DNA scelto, si ottiene cioè una eccezionale amplificazione. … e noi facciamo merenda..

23
10. Mentre nel termociclatore si amplifica il DNA si prepara l’ultimo passaggio che consiste nel l’elettroforesi del prodotto di amplificazione cioè il DNA ottenuto. Si riscalda del gel di agarosio per solubilizzarlo e versarlo nel contenitore (cella elettroforetica) dove, a temperatura ambiente il gel solidifica, si aggiunge anche un equivalente del bromuro di etidio per la lettura in fluorescenza. A questo punto con una sorta di “pettine” si scavano nel gel che sta solidificando i pozzetti in cui verrà deposto il DNA.
 dove, a temperatura ambiente il gel solidifica, si aggiunge anche un equivalente del bromuro di etidio per la lettura in fluorescenza. A questo punto con una sorta di pettine si scavano nel gel che sta solidificando i pozzetti in cui verrà deposto il DNA..")
24
11. Terminata la PCR ogni allievo riprende la propria provetta con il DNA amplificato. Gli allievi si alternano secondo il loro numero d’ordine per caricare con il proprio campione i pozzetti, uno per ogni allievo, della cella elettroforetica. La deposizione viene eseguita con estrema delicatezza considerando la consistenza del gel d’agarosio e la piccola quantità di DNA da deporre. Completata la deposizione si applica un voltaggio di 120 V per 30 minuti. Sotto l’azione del campo elettrico il DNA di ogni allievo viene separato sulla base del suo peso molecolare e migrerà dal polo negativo a quello positivo.

25
12. Alla fine della corsa elettroforetica è possibile visualizzare il DNA quando il gel viene illuminato con luce ultravioletta, grazie alla presenza del preparato per la lettura in fluorescenza.

26
13. La lettura dei risultati può evidenziare
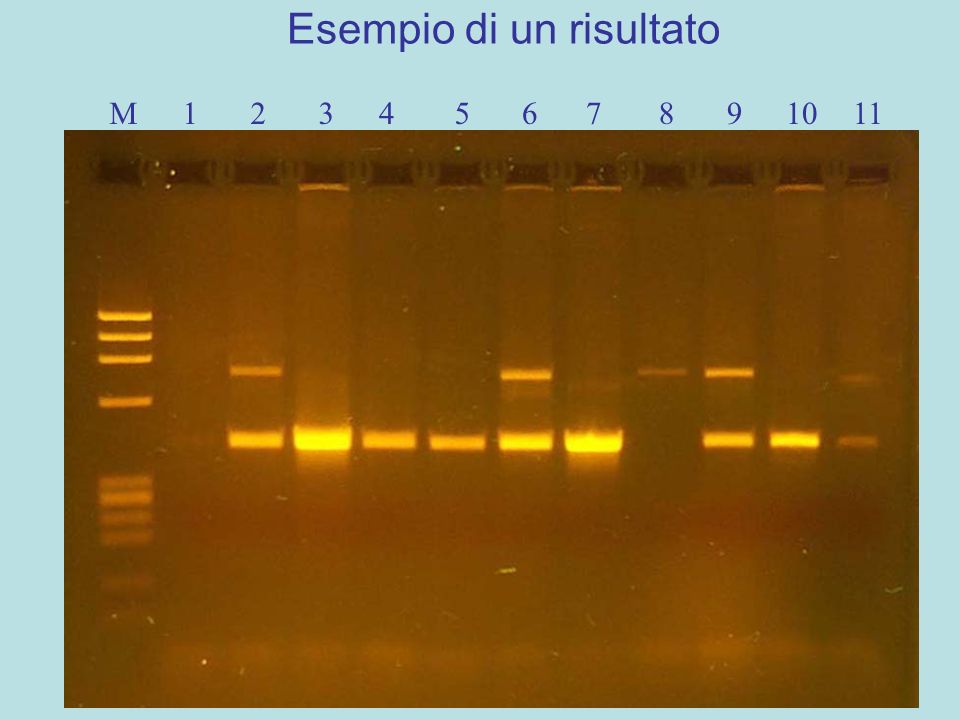
una sola banda visibile di 715 bp, PV92 contiene gli elementi Alu su entrambi gli alleli, omozigote per l’inserzione PV92 Alu (+/+) una sola banda visibile di 415 bp, gli elementi Alu sono assenti su entrambi gli alleli di PV92, omozigote per l’assenza dell’inserzione PV92 Alu (-/-) due bande visibili di cui una di 715 bp e l’altra di 415 bp, nel gene PV92 Alu è presente solo su un allele, eterozigote per l’inserzione PV92 Alu (+/-).
 una sola banda visibile di 415 bp, gli elementi Alu sono assenti su entrambi gli alleli di PV92, omozigote per l’assenza dell’inserzione PV92 Alu (-/-) due bande visibili di cui una di 715 bp e l’altra di 415 bp, nel gene PV92 Alu è presente solo su un allele, eterozigote per l’inserzione PV92 Alu (+/-).")
27
Esempio di un risultato
1 2 3 4 5 6 7 8 9 10 11
Presentazioni simili
















. In questo centro diviso.>")


